UKCA MHRA International Regulation Framework
MHRA Guidance update
On 21 May 2024, MHRA published a Statement of policy intent: international recognition of medical devices update.
This exciting development means that MHRA will start accepting medical devices that already meet EU, USA, Australian and Canadian regulations. There are, of course, conditions which must be met for manufacturers to be eligible for the necessary ‘certification of international recognition’.
We explain the succession of events that has brought us to this point and what the future holds for manufacturers entering the UK market.

MHRA Overview for UKCA
On 26 June 2022, MHRA published the government response to the public consultation on the future regulation of medical devices in the United Kingdom. The response outlined the intended regulatory reform including the transitional arrangements for CE marked devices placed on the Great Britain market which were put in place in 2023.
On 9 January 2024, the MHRA released a Roadmap towards the future regulatory framework for medical devices. This provides an update on the intended timelines to implement the future core regulations.
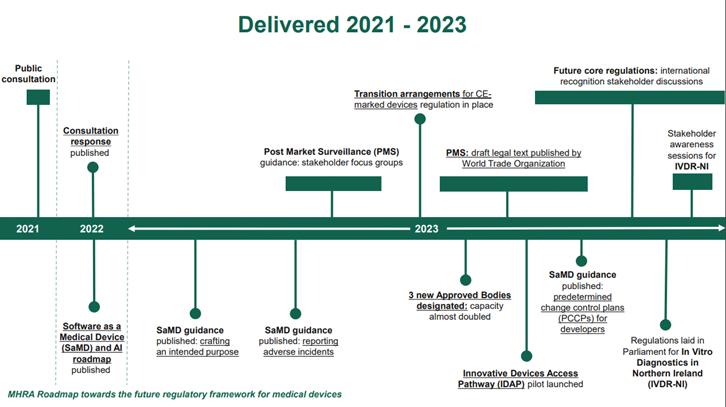
On 21 May 2024, following on from stakeholder discussions throughout the first quarter of 2024, MHRA released a statement of policy intent for recognition by the UK of approvals of medical devices from international regulators. This statement sets out MHRA’s proposed policy approach. The final legal text would come into force at the same time as the future core regulations.
UKCA Regulation Information
The UK government will introduce new regulations for medical devices. The intention is to prioritise patient safety, ensure patient access to the medical devices they need and keep the UK as an appealing market for MedTech.
The Government response to the 2021 consultation on the future regulation of medical devices in the United Kingdom detailed the intention to proceed with introducing alternative routes to market. This utilises approvals from other countries and Medical Device Single Audit Program (MDSAP) certificates in addition to the current UKCA (UK Conformity Assessed) marking process.
The UK currently recognises CE certification and approvals completed by the European Union (EU). This arrangement is time limited until 30 June 2030, at the latest, to support the transition following EU exit. The Medicines and Healthcare products Regulatory Agency (MHRA) also recognises other international regulators’ approvals for medicines.
As supported by the World Health Organization, reliance on the work of other regulators represents a ‘smarter’ form of regulatory oversight, resulting in more predictable, faster approvals to improve access to quality-assured medical devices for patients.
Additionally, reducing duplication of assessments conducted by comparable regulators will enable regulatory resource, and manufacturer resource, to be focused on more innovative products for the benefit of patient health.
Manufacturers of medical devices would still have the option to use the UKCA marking to place devices on the Great Britain market.
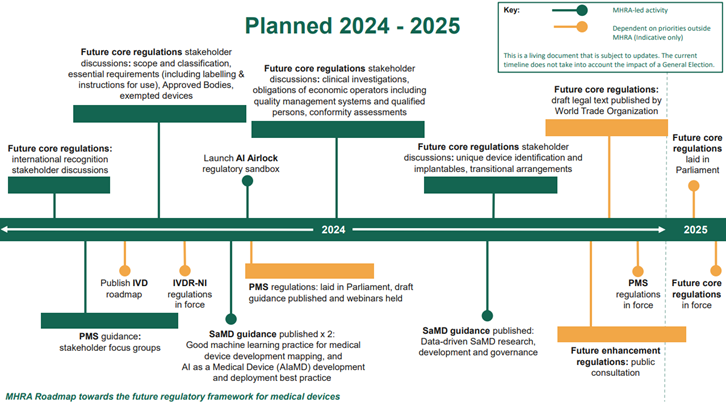
The regulations will be delivered through four statutory instruments. It is intended that priority measures to enhance post-market surveillance will be put in place first in 2024, with core elements of the new framework expected to be in place in 2025.
UKCA Regulation – What does this mean for your device?
International recognition will allow MHRA to utilise the expertise and decision-making of other regulatory partners for the benefit of patients. MHRA will retain the authority to reject applications if the evidence provided is considered insufficiently robust.
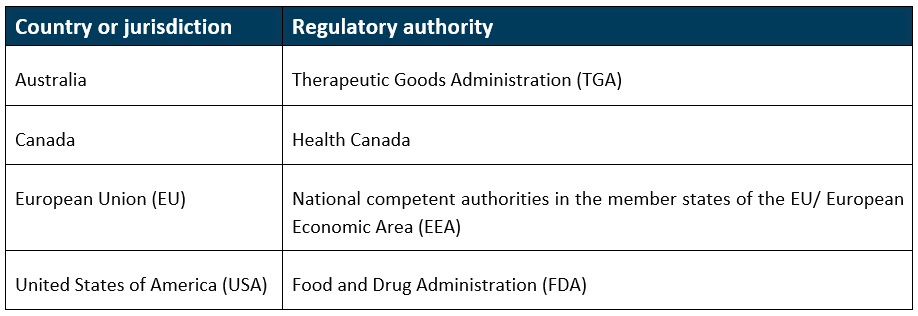
The comparable regulator countries (CRCs) for the proposed framework will be:
Is your device eligible?
To be eligible for the proposed framework, according to the proposed access routes described in Section 4, products will need to:
- comply with the relevant legislation in a CRC
- have English language labelling and packaging
- comply with Great Britain requirements for electronics compatibility (frequency, voltage and plug type), units of measurement, and labelling materials of concern where applicable (for example, for substances which are carcinogenic, mutagenic or toxic to reproduction (CMR), of category 1A or 1B, or could result in sensitisation or an allergic reaction)
- have all aspects of the device be in line with the device that is currently authorised in the CRC, including the design, manufacturing process and intended purpose
- have a UK responsible person, the name and address of which will be included on the label (this may be via over-labelling, and MHRA will also investigate the ability for digital labelling or digital label solutions)
- have a physical unique device identifier (UDI) on parts and labels in compliance with the requirements in the UK Medical Devices Regulations or the CRC
- comply with the new post-market surveillance (PMS) requirements in the UK Medical Devices Regulations which are expected to come into force in 2024
The proposed framework would enable device access to the UK market with a certificate of international recognition but would not provide a UKCA marking or UKCA certification.
Market access for eligible devices depends on the CRC certificate and must be re-certified when it expires.
If the CRC provides unlimited market access (as in the USA) the certificate of international recognition would be valid according to the quality management system (e.g., MDSAP) certificate.
What is excluded?
The following would be excluded from international recognition:
- exempted in-house devices
- custom-made devices
- Software as a Medical Device (SaMD) (including Artificial Intelligence as a Medical Device (AIaMD)) products that do not satisfy our intended purpose guidelines
- SaMD (including AIaMD) products approved via a route which relies on equivalence to a predicate (US 510(k))
- products granted market access in the CRC via a recognition route
- Class IIb (non-well established technology (WET)) implantable and III medical devices approved via a route which relies on equivalence to a predicate (US 510(k))
- companion diagnostics approved via a route which relies on equivalence to a predicate (US 510(k))
- companion diagnostics and combination products containing medicinal substances that are not licensed in the UK products excluded from the scope of UK MDR 2002, listed in The Medical Devices Regulations 2002, Regulation 3.
How will this framework operate?
This proposed framework is a draft, and the final version would come into force at the same time as the future core regulations which are aiming to be in force in 2025.
The operational aspects of this framework – who the documentation is submitted to, who reviews it, who performs post market surveillance and change assessment, and the requirements for these assessments – are being developed under trusted advisor principles in stakeholder discussions with industry, designated approved bodies and applicant approved bodies going through the designation process.
MHRA is testing the proposed framework using devices across all classifications and types to establish the process and associated guidance. Market access via international recognition would only be provided after the future core regulations are in force and the proposed framework may be changed accordingly.
The UK government wants to allow UKCA marked devices to be sold until the certificate expires or until 3 years after the new regulations take effect (for general medical devices) or 5 years (in the case of IVDs), whichever comes first.
The caveats that will apply to this arrangement are:
- devices that are subject to significant changes in design or intended purpose will be excluded from these provisions
- all post-market requirements applicable to the new regulatory framework will need to be complied with for all products which benefit from the transitionary arrangements

Proposed Access Routes
The classifications listed in each proposed access route refer to the classification that will apply under the UK Medical Devices Regulations, which may not be the same as the classification in the CRC.
Recognition, self-registration with MHRA
This applies for the following devices that comply with devices legislation in any of the specified CRCs (Australia, Canada, EU or USA):
- Class I medical devices, other than Class Is/m/r (Is: sterile; Im: measuring; and Ir: reusable)
- Class A IVDs that are non-sterile
A declaration by the manufacturer to an appropriate quality management system such as ISO 13485 or product-specific equivalent will be required for these devices.
Reliance
This applies for the following devices, excluding AIaMD (Artificial Intelligence as a Medical Device) and devices where the classification is different in the EU to that under the UK Medical Devices Regulations, that comply with devices legislation in the EU[1]:
- Class Is/m/r medical devices that comply with EU Medical Devices Regulation (EU MDR)
- Class IIa, IIb, III medical devices that comply with EU Medical Devices Regulation (EU MDR)
- sterile Class A IVDs that comply with EU In Vitro Diagnostic Medical Devices Regulation (EU IVDR)
- Class B, C, and D IVDs that comply with EU In Vitro Diagnostic Medical Devices Regulation (EU IVDR)
Manufacturers will need to submit a dossier in the International Medical Device Regulators Forum (IMDRF) ‘table of contents’ format, or equivalent format used in the CRC.
Evidence of approval and a PMS plan and associated PMS report or periodic safety update report (PSUR) will be reviewed.
For AIaMD, or devices where the classification is different in the CRC to that under the UK Medical Devices Regulations, please see ‘Reliance with abridged assessment and device-specific requirements’ below.
Reliance with device-specific requirements
This applies for the following devices, excluding AIaMD and devices where the classification is different in the CRC to that under the UK Medical Devices Regulations, that comply with devices legislation in Australia or premarket approval legislation in the USA:
- Class Is/m/r medical devices
- Class IIa, IIb, III medical devices
Manufacturers will need to submit a dossier in the IMDRF ‘table of contents’ format, or equivalent format used in the CRC.
Evidence of approval and a PMS plan and associated PMS report or PSUR will be reviewed. Manufacturers will need to supply implant cards and patient information leaflets for implantable devices for the UK. A summary of safety and clinical performance (SSCP) will also be needed for Class III and implantable devices.
For AIaMD, or devices where the classification is different in the CRC to that under the UK Medical Devices Regulations, please see ‘Reliance with abridged assessment and device-specific requirements’ below.
Reliance with abridged assessment and device-specific requirements
This applies for the following devices:
- Class Is/m/r medical devices that comply with devices legislation in Canada or DeNovo or Premarket Notification (510k) legislation in the USA
- Class IIa, IIb, III medical devices that comply with devices legislation in Canada or DeNovo legislation in the USA
- Class IIa, IIb (non-implantable) and IIb (well established technology (WET)) medical devices that comply with 510k legislation in the USA
- sterile Class A IVDs that comply with devices legislation in Australia, Canada or Premarket Approval, DeNovo or 510k legislation in the USA
- Class B, C, and D IVDs that comply with devices legislation in Australia, Canada, or Premarket Approval, DeNovo or 510k legislation in the USA
- AIaMD (Artificial Intelligence as a Medical Device) that comply with devices legislation in Australia, Canada, EU[2], or Premarket Approval or DeNovo legislation in the USA
- any device where the classification is different in the CRC to that under the UK Medical Devices Regulations
Manufacturers will need to submit a dossier in the IMDRF ‘table of contents’ format, or equivalent format used in the CRC.
Evidence of approval and a PMS plan and associated PMS report or PSUR will be reviewed.
Device classification will be reviewed since this route captures devices with different CRC and UK Medical Devices Regulations classifications. Manufacturers must supply implant cards and patient information leaflets for implantable devices. A summary of safety and clinical performance (SSCP) is needed for Class III and implantable devices, and Class C and D IVDs. Clinical data may be sampled, and AIaMD will review premarket (training and test) data, implementation verification and validation and use of predetermined change control plans.
CLIN-r+ recommendations
UKCA remains a valid path to the UK market. Even with the new recognition system, the UKCA mark will still be accepted. The MHRA plans to allow a transition period of 3 years for medical devices and 5 years for IVDs for those holding the UKCA mark.
The MHRA is still in the testing phase and aims to bring this policy into force in 2025. However, manufacturers should start planning early and look at eligibility at application requirements to determine the best route.
Some devices, like custom-made devices and certain medical software, will be excluded. The MHRA plans to finalize this recognition approach by 2025 as part of the new UK medical device law. Until then, discussions and testing will define specific elements, like documentation review and post-market surveillance oversight.
It’s worth noting that Northern Ireland has remained part of the European single market for medical devices post-Brexit. Therefore, Northern Ireland requires CE Marking and follows EU regulatory requirements.
We advise you obtain to professional assistance to evaluate your application and technical file. Clin-r+ can advise and support you to compile and provide the specific requirements needed for eligibility. We can assist with IMDRF compliant dossiers and help you gain a certificate or international recognition that will grant access to the Great Britain market. CLIN-r+ has a wealth of experience to call upon. Get in touch!
