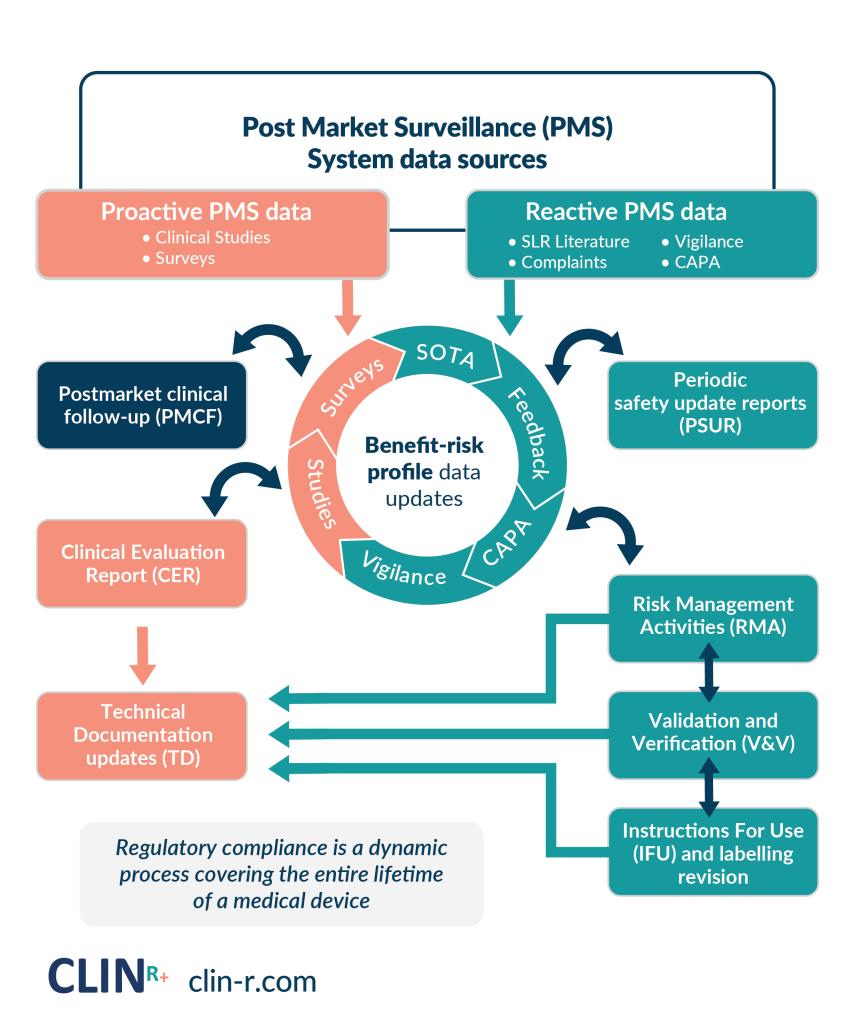
Don’t have the in-house resources or expertise to have realtime PMS data or automate your PMSR/PSUR!
Your current PMS plan and PSUR had deficiency findings in your current audit.
Are you unsure if your current PMSR/PSUR will meet the EU MDR requirements?
Your legacy PSUR has had deficiency findings in one or more of your total 3 reviews with your notified body.