Clinical Investigation Exemptions
Do manufacturers need to conduct investigations or follow an exemption route?
The conformity assessment route according to the EU MDR classification is well known. However, there is something that’s often overlooked. A lot of manufacturers delay the formulation of the Clinical Development Plan (CDP). Therefore, the clinical evaluation conformity route based on the device history, novelty, and current clinical data available is missed. The CER is the biggest source of non-conformances and a leading cause of CE marking delays. But worse still is investing in clinical investigation when it’s not needed.
The clinical evaluation conformity routes can be split into two categories: those that are exempt from Clinical Investigations and those that are not. There are 4 exceptions as outlined by MDCG 2023 and MDCG 2020-6. This paper looks to explore how the CDP selects the Clinical Evaluation conformity routes and explores each route.

Exempted Devices - Article 61(6b)
If you have an exempted device then you need to follow MDR Article 61(6). This states:
“The requirement to perform clinical investigations pursuant to paragraph 4 shall not apply to implantable devices and class III devices:
(a) which have been lawfully placed on the market or put into service in accordance with Directive 90/385/EEC or Directive 93/42/EEC and for which the clinical evaluation:
- is based on sufficient clinical data, and
- is in compliance with the relevant product-specific CS for the clinical evaluation of that kind of device, where such a CS is available; or
(b) that are sutures, staples, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips or connectors for which the clinical evaluation is based on sufficient clinical data and is in compliance with the relevant product-specific CS, where such a CS is available.”
Any device on the list in Article 61(6b) that also meets the criteria in MDCG 2020-6 may be considered as Well Established Technology (WET).
Devices grouped under WET may be able to establish conformity with the relevant GSPRs by evaluating cumulative clinical data evidence such as:
- Equivalence data.
- State of the Art (SOTA).
- Vigilance data (including complaints).
- Proactive Post Market Surveillance (PMS) data.
- Case reports on the target device.
- Compliance with non-clinical parts of common specifications relevant to device safety and performance.
- Simulated use data.
- Pre-clinical and bench testing/ compliance with standards.
It should be noted that clinical evidence only based on PMS data will not be considered sufficient. The device classification will also have a significance as to how much clinical evidence is required.
Medical Devices where Clinical Data not appropriate - Article 61(10)
In this case, you need to follow MDR Article 61(10). This states:
“Without prejudice to paragraph 4, where the demonstration of conformity with general safety and performance requirements based on clinical data is not deemed appropriate, adequate justification for any such exception shall be given based on the results of the manufacturer’s risk management and on consideration of the specifics of the interaction between the device and the human body, the clinical performance intended and the claims of the manufacturer. In such a case, the manufacturer shall duly substantiate in the technical documentation referred to in Annex II because it considers a demonstration of conformity with general safety and performance requirements that is based on the results of non-clinical testing methods alone, including performance evaluation, bench testing and pre-clinical evaluation, to be adequate.”
Here, clinical evaluation relies solely on pre-clinical data. The evidence required is:
- Compliance with non-clinical parts of common specifications relevant to device safety and performance.
- Simulated use data.
- Pre-clinical and bench testing/ compliance with standards.
In this case, a strong justification needs to be provided by the manufacturer. It must be clear why the demonstration of conformity with GSPRs based on non-clinical testing methods alone is adequate. Justification for this should be based on:
- Risk management results.
- The intended clinical performance of the device.
- Interaction between the device and the human body.
- Manufacturer claims.
SoTA supports that clinical data is not appropriate is helpful.
Legacy Devices registered under MDD - Article 61(6a)
If the legacy device has sufficient data, then you should follow Article 61(6a).
However, if the device meets WET and is in the range of the SOTA benchmark then MDCG 2020-6 should be followed. This guides manufacturers on the clinical evidence needed for medical devices previously CE marked.
“Sufficient clinical evidence” isn’t defined in the MDR. However, it is understood as “the present result of qualified assessment which has concluded that the device is safe and achieves the intended benefits.” It’s important to note that clinical evaluation is a process where the ‘qualified assessment’ must be done continually.
The level of clinical evidence required for the device needs to be determined by the manufacturer and verified by the notified body. It must be appropriate given the characteristics of the device and its intended purpose. This should include:
- WET checklist.
- SOTA data.
- Clinical evaluation Report (CER) data gap analysis.
- PMCF study.
Claiming Equivalence - Article 61(5) and (4)
Here, manufacturers should check if the device meets the requirements in Table 2 of MDCG 2023-7 (below). Manufacturers must show sufficient access to device data to justify equivalence. For example, manufacturers may have the latest technical sheet and routinely test/characterize equivalent devices.
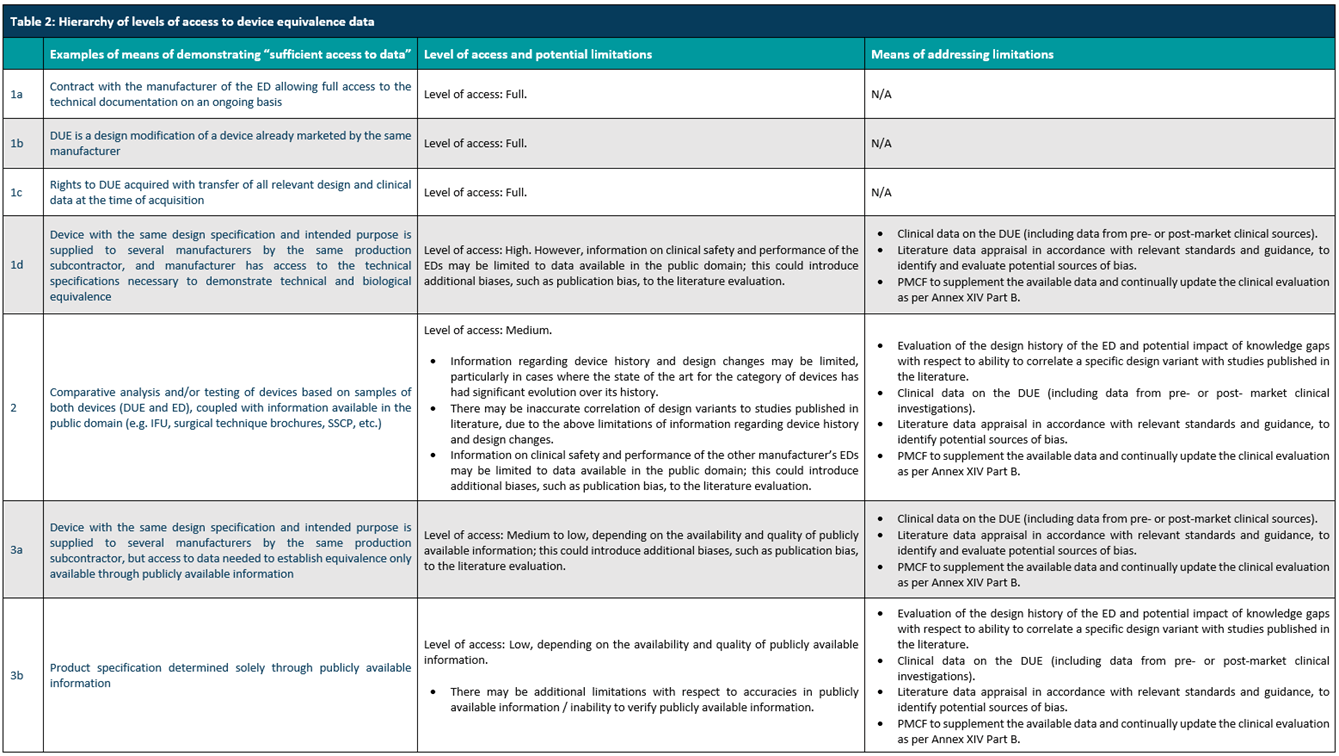
To show equivalence to another device, manufacturers must demonstrate and justify the equivalence based on clinical, technical, and biological characteristics. Studies should be conducted and documented to show technical and biological equivalence. MDCG 2020-5 guides to perform equivalence justification. Again, the level of clinical evidence deemed sufficient is proportionate to the device classification. This should include:
- Equivalence justification.
- Technical equivalence studies.
- Biological equivalence studies.
- PMCF study.
Conditions where a Clinical Investigation is needed - Article 61(3) and Annex XV
There are still situations here where Clinical investigation is required. This is the case if the device is either:
- Pre-market.
- Without clinical data.
- A novel device that’s not WET.
The EU MDR outlines general requirements for conducting clinical investigations and gives details on requirements such as:
- Methods.
- Documentation regarding the application for clinical investigation.
- Clinical Investigation Plan.
- Obligations of the sponsor.
In order to conduct a successful clinical investigation, several elements should be considered and carried out. Clinical investigation should be planned in the Clinical Development Plan (CDP) however a separate Clinical Investigation Plan (CIP) is still required. Parameters must be planned through State of the Art (SOTA). A Statistical Analysis Plan (SAP) must be developed and a Gap Analysis that meets ISO 14155 standards should be carried out.
The Clinical Investigation Report (CIR) must also meet MDCG 2020-6 criteria for sufficient clinical data.
CLIN-r+ recommendations
When clinical data is insufficient to cover all devices, intended use, safety, and performance claims, manufacturers should conduct a gap analysis based on MDR standards. Data gaps can be filled in several ways. PMCF studies may be the best way to gather clinical evidence, although scientifically sound questionnaires or registries can also be used. Manufacturers might limit the device’s intended use until clinical data is available.
Where the manufacturer provides no or an ill-defined SoTA, it opens the door for the Notified Body to impose their interpretation of the SoTA. Uncertainty may need costly clinical trials or no CE mark. Defining the ‘state of the art’ should be the starting point in the development process by any manufacturer or innovator in MedTech.
A state of the art (SoTA) literature review gives a wealth of information. It overviews the performance and safety data, and defines the technology’s clinical benefits, disadvantages, risks, and limitations. It contains powerful strategic insight on whether a clinical investigation is needed, what type of design would be best to address your gaps, help support your ‘Intended Purpose’ and help keep your trial budget in check.
Manufacturers without this expertise should consider partnering with an experienced consultancy when assessing standards, risk, and clinical data. Or considering what clinical studies are needed for your devices’ CE marking needs. Consultancies also have resources such as clinical study designers, statisticians and experienced SoTA medical writer’s systematic review helping expand your companies’ capabilities cost-effectively.
Clin-r+ provides expert assistance and has a wealth of experience to call upon. We can assist with your transition and ensure you are MDR compliant. To learn more about our services and how we can help. Get in touch
