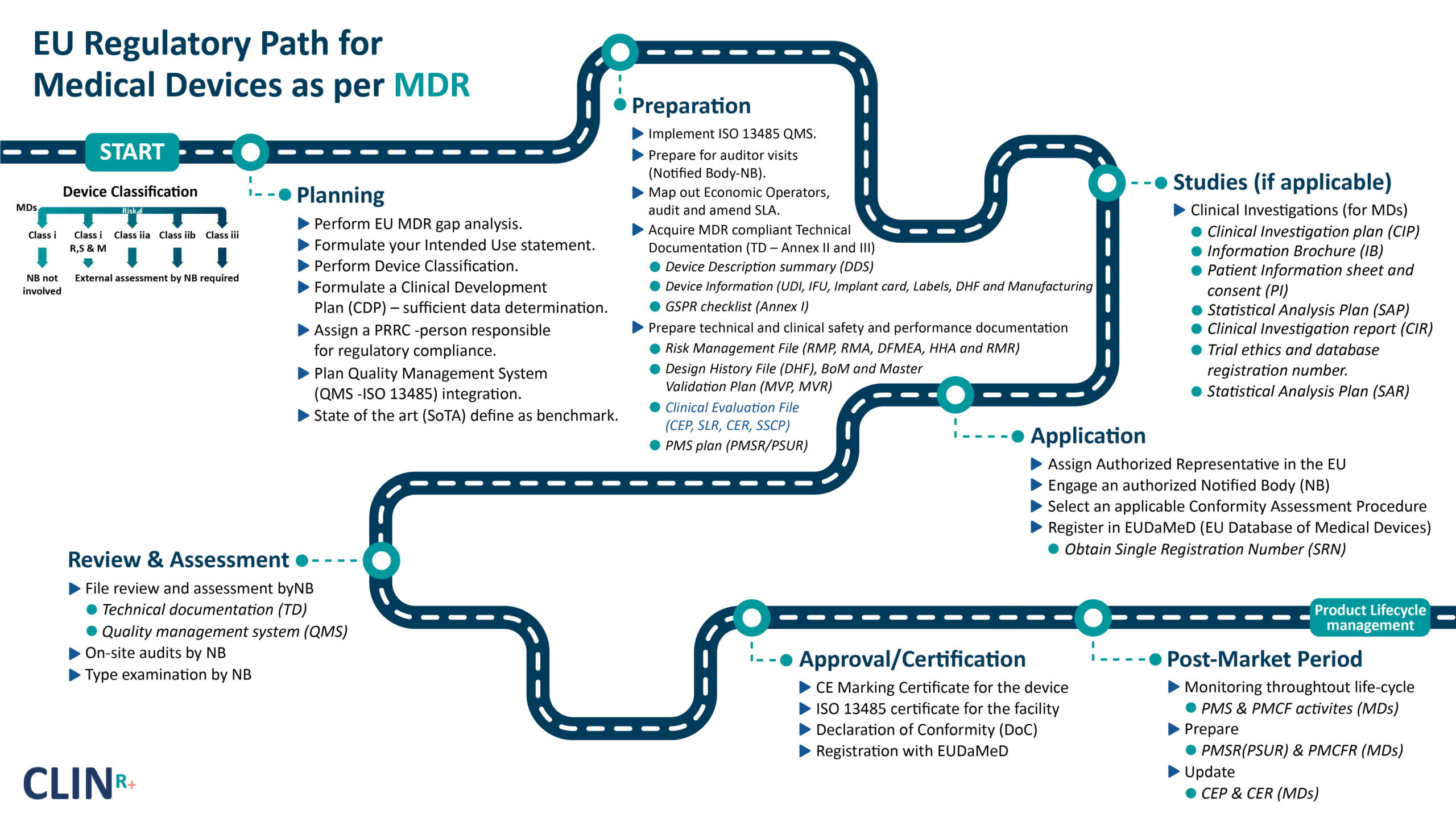
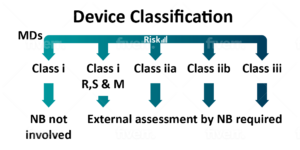
Devices are segregated further into different classifications (listed below).
IVDs have their own classification system. IVD companies can also expect Notified Bodies to be more involved in the regulatory process than previously.
Under MDR, active implantable devices are still subject to similar requirements as Class III devices.
- Class I (low risk).
- Class Is (product that is delivered sterile).
- Class Im (product with a measuring function).
- Class Ir (products that are reprocessed).
- Class IIa (medium risk).
- Class IIb (medium/high risk).
- Class III (high risk).
