Manufacturers of class III and implantable medical devices may avoid clinical examination under MDR Article 61(4) if they can establish the following equivalence:
- modifications of a device already marketed by the same manufacturer have designed the device,
- equivalence can be demonstrated according to the MDR between the devices, and
- the clinical evaluation of the marketed device is sufficient to demonstrate conformance with the applicable safety and performance requirements.
Article 61(4) of Regulation (EU) 2017/745 on medical devices (MDR) requires clinical investigations to be performed for implantable and class III devices, except in four specific cases as outlined in: CASE 1) indents 1-3 of Article 61(4); CASE 2) Article 61(6)(a); CASE 3) Article 61(6)(b); CASE 4) Article 61(5) in MDCG 2023-7.
According to MDR Article 61(5), the manufacturer of implantable and class III devices that claim to be equivalent to a product already commercialised, but not made by the same company, must have a contract to grant continuing access to the product’s technical and clinical documentation. In this case, the equivalent device must have passed its initial clinical examination in accordance with MDR and be MDR certified. According to the MDCG 2020-5, MDD-certified devices cannot be used to assert equivalence.
According to the MDCG 2023-7, provides a summary of the cases when implantable and class III devices may be exempted from mandatory clinical investigations.

CASE 1:
Indents 1-3 of Article 61(4)
• DUE has been designed by modifications of a device already marketed by the same manufacturer.
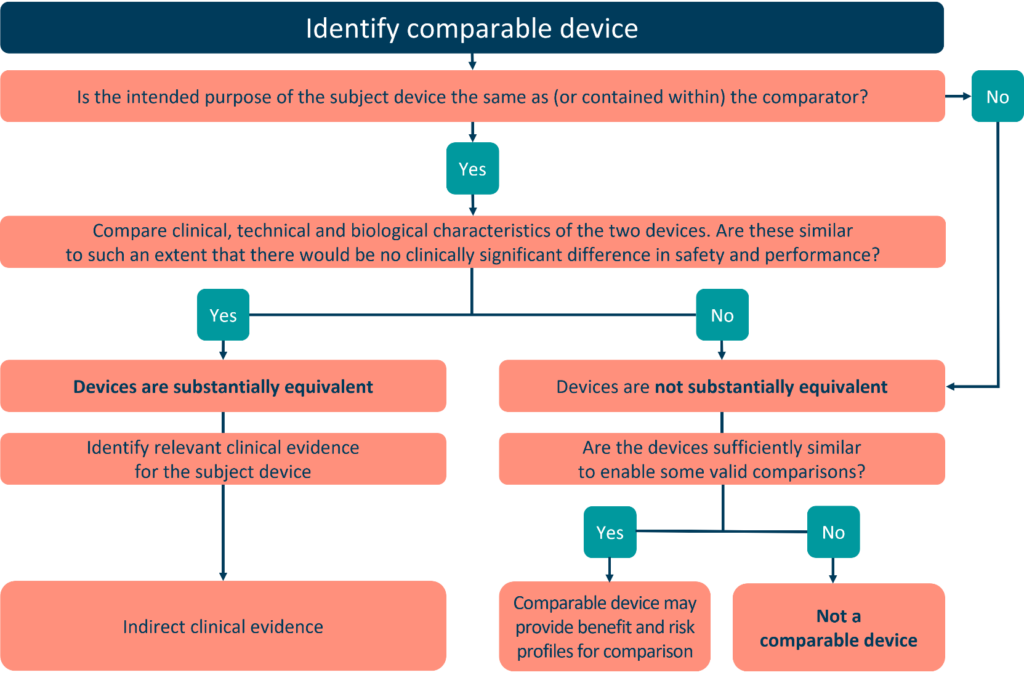
• Equivalence is demonstrated between the DUE and the manufacturer’s ED in accordance with Section 3 of Annex XIV; demonstration of equivalence has been endorsed by the notified body. For further guidance on the demonstration of equivalence,
please refer to MDCG 2020-5.
• The clinical evaluation of the marketed device is sufficient to demonstrate conformity of the modified device with the relevantsafety and performance requirements4, 5
• PMCF plan is appropriate and includes post market studies to demonstrate the safety and performance of the DUE6
CASE 2:
Article 61(6)(a)
• DUE has been lawfully placed on the market or put into service in accordance with Directive 90/385/EEC or Directive 93/42/EEC.
• The clinical evaluation is based on sufficient clinical data
• The clinical evaluation is in compliance with the relevant productspecific CS for the clinical evaluation of that kind of device, where such a CS is available.
CASE 3:
Article 61(6)(b)
- DUE is one of the listed types of devices: “sutures, staples, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips or connectors”.
- The clinical evaluation is based on sufficient clinical data
- The clinical evaluation is in compliance with the relevant product specific CS for the clinical evaluation of that kind of device where such a CS is available.
CASE 4:
Article 61(5)8
- Equivalence is demonstrated between the DUE and the other manufacturer’s ED in accordance with Section 3 of Annex XIV; demonstration of equivalence has been endorsed by the notified body (via Article 61(4)).
- The clinical data from the clinical evaluation of the ED is sufficient to support the intended purposes of the DUE (via Article 61(4))9
- The two manufacturers have a contract in place that explicitly allows the manufacturer of the ED full access to the technical documentation on an ongoing basis.
- The clinical evaluation of the other manufacturer’s ED has been performed in compliance with the requirements of the MDR10, 11
- PMCF plan is appropriate and includes post market studies to demonstrate the safety and performance of the DUE6 (via Article 61(4)).