Clinical Evaluation for Medical Devices
What you need to know for EU MDR
MEDDEV 2.7/1 Rev 4 was released in 2016 as a Clinical Evaluation guide for manufacturers and notified bodies. They announced the EU MDR in May 2017. The MDR affirms the MEDDEV and provides further information on the clinical data requirements. We explain the key stages of clinical evaluation and what documentation we need in this white paper.
What is Clinical Evaluation?
MDR defines clinical evaluation as “a methodologically sound ongoing procedure to collect, appraise, and evaluate if there is sufficient clinical evidence to confirm compliance with the relevant essential requirements for safety and performance when using the device according to the manufacturer’s Instructions for Use”.
The clinical evaluation must demonstrate the safety and performance of the device.
The requirements for clinical evaluation apply to all classes of medical devices. The evaluation should be appropriate to the device under evaluation, its specific properties, and its intended purpose. We should specify benefits and risks, e.g., their nature, probability, extent, duration, and frequency. Core issues are the proper determination of the benefit/risk profile in the intended target groups and medical indications, and the demonstration of acceptability of that profile based on current knowledge/ the state of the art in the medical fields concerned.
Clinical evaluation is the responsibility of the manufacturer, and the clinical evaluation report is an element of the technical documentation of a medical device. It is conducted throughout the life cycle of a medical device, as an ongoing process.
What are the 5 phases of Clinical Evaluation?
According to MEDDEV, the clinical evaluation consists of 5 stages:
0 – Define the scope and plan the evaluation.
1 – Identify pertinent data.
2 – Appraise each individual data set in terms of its scientific validity, relevance and weighting.
3 – Analyse the data and reach conclusions.
4 – Finalise the Clinical Evaluation Report (CER).
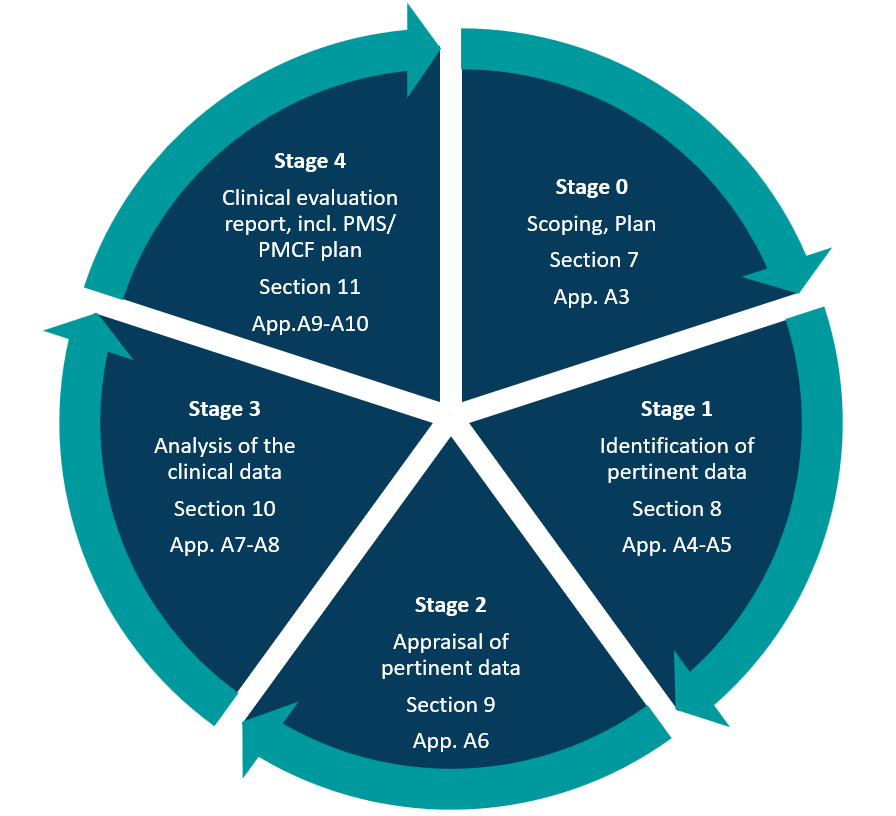
Stage 0 – Defining the scope
Stage 0 scopes the clinical evaluation and plans each step. MEDDEV 2.7/1 Rev 4 requires a documented Clinical Evaluation Plan (CEP) at this stage. MEDDEV Section 7, Appendix 3 emphasizes the importance of a CEP and outlines details. The CEP defines the scope and clinical strategy of the clinical evaluation, considering the device’s type and history.
The CEP determines if the CER will use equivalence (the test and reference devices are similar to the extent that there would be no clinically significant difference in the safety and clinical performance of the test device) and/or clinical data and the types of clinical data presented (ie, bench-top testing, clinical studies, or literature). It will also reference risk assessments, IFUs, literature, and PMS databases. It should be updated as needed when the device, current knowledge/state of the art, applicable standards/guidance documents, medical condition managed by the device, or medical alternatives change.
The CEP needs a device overview and must describe the device, its design, indications, intended use, warnings, potential complications, and target population. The manufacturer should also make clinical performance and safety claims. This section covers regulatory approvals and changes since the last report.

Stage 1 – Identification of pertinent data
Stage 1 identifies and gathers the data for review as part of the clinical evaluation. Clinical data includes preclinical studies, clinical studies, and clinical literature. A literature search protocol (LSP) must identify and describe clinical literature. The MEDDEV states that an LSP should clearly define the methodology and sound strategy for the search, and provide examples of methods such as:
- PICO (patient characteristics, type of intervention, control, and outcome queries)
- PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses)
- MOOSE (meta-analysis of observational studies in epidemiology)
The LSP should comprise search methodology, literature sources, database search strategy, internet search extent, search terms and limits, timeframe, inclusion/exclusion criteria, and appraisal plan. We can search scientific databases, internet searches, non-published data, and scientific literature citations for data. MEDDEV 2.7/1 Rev 4 Appendix 4 recommends MEDLINE or PubMed. We can utilise Embase, Excerpta Medica, and the Cochrane CENTRAL trials register to cover devices used in the EU.
Post-market performance data should include manufacturer complaint and recall data, MAUDE data (US), BfArM (Germany), Healthy Canadians (Canada), Swiss Medic (Switzerland), and other relevant databases like Eudamed (when available). New devices need to include all data. Manufacturers need to coordinate with the notified body and have enough data to show post-market trends for existing device updates. Data identification must be thorough, verifiable, and justify inclusion and exclusion.
Stage 2 – Appraisal of the pertinent data
Stage 2 appraises the data found. We must assess data for inclusion or deletion, depending on its contribution to device safety and performance. The appraisal process should evaluate each document’s quality, validity, and relevance. Next, we must systematically weigh it for contribution to the clinical evaluation using a systematic approach (Figure 1). Transparent appraisals should contain all high-quality data, positive or negative.
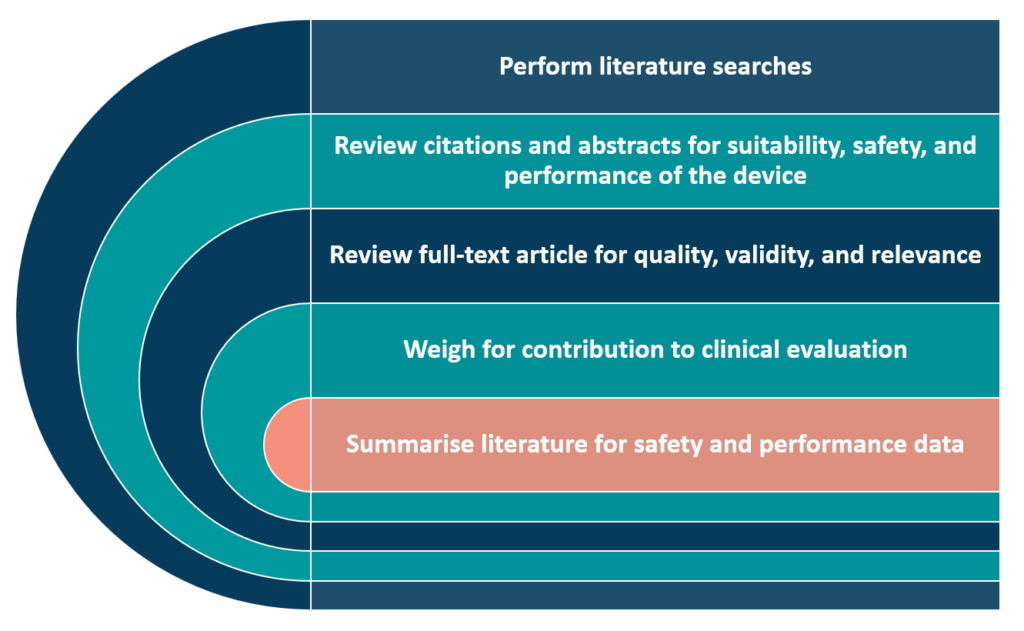
Include an appraisal plan with the methodological selection criteria to ensure unbiased clinical data evaluation. MEDDEV Appendix 6 lists studies that lack scientific validity for clinical performance and safety. Excluded papers omit techniques, products, patients, clinical outcomes, confidence intervals, or statistical significance. Data quantity may determine inclusion. Some devices have hundreds of meta-analyses and randomised clinical trial data, while others have only single-centre observational studies.
Stage 3 – Analysis of the clinical data
Stage 3 assesses conformity requirements. Previously, with the essential requirements (ERs) 1, 3, and 6 of the MDD and now for general safety and performance requirements (GSPRs) 1, 5, and 8 of the MDR:
- ER 1 = GSPR 1 and 5
- ER 3 = GSPR 1
- ER 6 = GSPR 8
Data should show that clinical evidence meets all of the above requirements.
The clinical data for each requirement must be analysed to determine that the device is designed and manufactured to ensure patient and user safety. It should also meet the “state of the art” (SOTA) products that are developed and approved for sale in the marketplace. To ensure relevant standards address all identified hazards and any gaps are covered by clinical data, we should review the data using risk management documents.
The manufacturer must correctly identify the medical conditions and target groups the device will treat and demonstrate clinical evidence of the patient benefits. The data should also show an acceptable benefit/risk profile for the intended purpose. Patient risks must be minimised and acceptable when weighed against patient benefits.
Any residual risks, uncertainties and concerns should define post market clinical follow up needs.
Stage 4 – Clinical evaluation reporting
Stage 4 concludes the clinical evaluation by documenting each phase and assessing the device’s safety and performance. For more information, consult the MEDDEV’s sample table of contents in Appendix 9. Below is a table of contents summary.
- Summary: This is a summary of the report and should include the guidance being conformed to (MDR), a benefit/risk profile, target groups, medical indications, and device profile acceptability based on the state of the art.
- Scope of the clinical evaluation: The manufacturer must list information and provide images for the device, accessories, name/address, and physical and chemical characteristics. Also include IFU information such as indications, warnings and complications.
- Clinical background, current understanding and state of the art: This should include a literature search including the methodology, sources and appraisal criteria. State of the art information is crucial in justifying the device in its intended market. You should describe similar and competitive devices here along with their risks and benefits. Along with a description of the medical condition, population, alternatives to the device being used, users and unmet medical needs should also be included.
- The device under evaluation: we must state the type of evaluation, along with information on what it’s based on, demonstration of equivalence (if applicable), and the conclusion(s).
- Conclusions: This section of the CER should state compliance to the MDR with the acceptability of the benefit/risk profile, according to both current knowledge and state of the art. The manufacturer must also list how they will follow up and report on the finding post market.
- The next clinical evaluation date and how it was determined based on the device class and how well established it is.
Key considerations
We advise taking some key questions into consideration when undertaking your clinical evaluation:
- Are your clinical evaluation reviewers “qualified”?
- Do you have all evaluators’ CVs and declarations of interest?
- Have you searched multiple databases and methodologically explored clinical literature for state of the art and clinical data?
- Do the report’s findings have “sufficient” clinical evidence?
- Are conformity to all GSPRs stated and inconsistencies noted?
- Do you have a Clinical Evaluation Plan (CEP), A Post Market Surveillance (PMS) Plan, and a Post Market Follow Up (PMCF) Plan?
CLIN-r+ Recommendations
We know that the new regulations and all that clinical evaluation now entails can be problematic. Clinical Evaluation has evolved from a simple process and report to a detailed justification of your medical device and critical review of its safety and performance. Data from trials and comprehensive literature searches must support the evaluation.
Clinical evaluation, including the CEP, CER, literature searches and report, is a key component of the MDR Technical Documentation, which must be done to obtain CE marking.
Should you have any questions or need professional assistance, CLIN-r+ has a wealth of experience in clinical evaluations and literature searches to call upon. Get in touch!
