Clinical Investigations for Medical Devices and IVD
What are the requirements?
The EU MDR defines clinical investigations as “any systematic investigation involving one or more human subjects, undertaken to assess the safety or performance of a device.”
Clinical data (data obtained from use in humans) is a requirement for CE Marking and EU marketing of all Classes of Medical Devices and Invitro Diagnostic tests. Clinical studies (real-patient studies) must meet various regulatory standards before starting.
The EU MDR requires companies to develop a ‘Clinical Development Plan’ (CDP) to map out pre-clinical and post-market activities to obtain clinical data. This should be the starting point where manufacturers centralise departmental goals for the product and plan clinical investigations to meet Good Clinical Practices (GCP’s), ICH guidelines, and the regulations that govern clinical trials in the parts of the world they want to market in, such as EU MDR or the US FDA 21 CFR Parts 50, 56, and 812.
The CDP should intentionally align all clinical investigations on the international standard of medical devise for human subjects (ISO 14155) which will assure that as well as European guidance documents such as MEDDEV 2.7/4. This white paper outlines the requirements and considerations manufacturers must review to ensure well-researched clinical investigations and a return on investment.
Clinical Investigation regulatory pathways in the EU – MDCG 2021-6 guidance
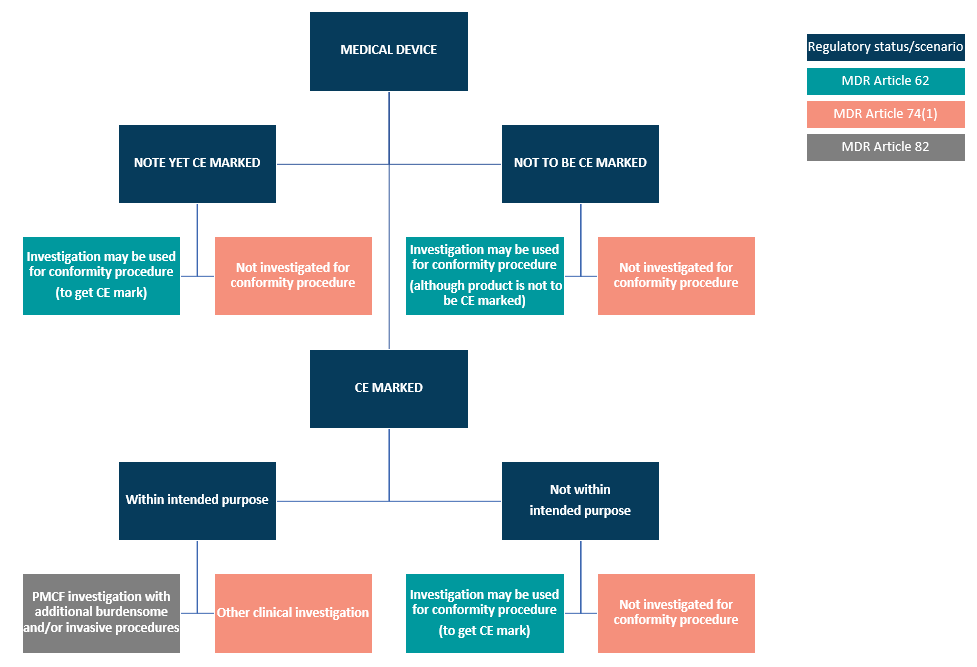
EU MDR has 20 articles that outline the requirements for clinical investigations of medical devices, spanning articles 62 through 82. Within these articles, the regulation lays out three regulatory pathways manufacturers can take:
- Article 62 covers clinical investigations to confirm compliance and get a CE marking. This is the pathway medical device companies will use if their device classification (for Class III or Class IIb implantables) requires a clinical investigation.
- Article 74(1) regulates CE-marked devices if the investigation parameters are within the device’s intended purpose. In other words, if you are conducting a clinical investigation as part of your Post-Market Clinical Follow-Up (PMCF), then Article 74(1) will then guide you.
- Article 82 covers clinical investigations that are not being performed in order to demonstrate conformity. Additionally, the Member State in which you hold your study may have relevant national provisions for you to follow.
To clarify the routes MDCG 2021-1 illustrates this pathway but draws attention to national guidelines that should be reviewed.
Hence, be clear when planning your CDP what regions you’re considering trials in and why. Consider the long term needs for clinical data as the MDR states this is an ongoing process throughout the devices’ lifetime. Plan carefully using your SoTA to avoid expensive trials just because a CRO convinced you.
Stages and types of medical device clinical Investigations
We can carry out Clinical investigations (trials) during both the premarket and post-market phases of the device lifecycle. Medical device manufacturers may conduct several clinical activities, including clinical trials, during pre-market and post-market phases, as illustrated below.

Clinical investigations/trials may occur during the pilot stage, the pivotal stage, or during post-market surveillance.
As previously mentioned, pre-clinical activities do not use human subjects, but this is not the same as premarket pilot and pivotal studies.
Several study descriptions are interchangeable and used in different markets to describe the same thing. So, let’s focus on the general types and stages of studies and their burden to human subjects.
Pilot studies commonly precede gadget design. Pilot studies evaluate device functionality and clinical safety, because nonclinical testing cannot. They involve 10 or fewer patients. Usability studies, which may accompany a pilot study, are different.
Pilot studies gather a wide range of data to:
- Identify modifications to the device or procedure
- Optimize operator technique
- Refine the intended use population
- Refine nonclinical test plans or methodologies
- Develop subsequent clinical study protocols
When SoTA data is insufficient and raises questions about safety and performance endpoints, pilot study data can help design a pivotal study. A pivotal study is used to gather definitive evidence of the safety and effectiveness of your medical device for a specific intended use. These studies generally use a larger number of subjects than pilot studies, and you’ll use the results of your pivotal study to gain regulatory approval for your device.
A pivotal study does not necessarily need to be preceded by a pilot study. This is where a well written SoTA can save time and money in developing your device. If your systematic review of the SoTA is comprehensive, it will provide suitable safety and performance endpoints. It will also provide prior art (reference range of your performance and safety) effect size to determine study sample size.
The types of clinical investigations you carry out will depend on your device and the regulatory pathway you’re taking.
Unlike drug studies, the guidance does not recommend a study design. Meaning you can utilise your SoTA data if you can justify well-established technology and your CE position in markets outside the EU to obtain observational post-market clinical data to help your EU MDR clinical data needs.
Do clinical trials happen during post-market surveillance?
As you can see from the graphic, the post-market surveillance stage includes both confirmatory and observational types of clinical activities. While it may seem odd that you would need to perform a confirmatory study after receiving approval to place your device on the market, this is not an irregular occurrence. For example, EU MDR includes a distinct regulatory pathway – Article 74(1) – for conducting a clinical investigation as part of your PMCF.
Post-market surveillance studies can confirm the device’s safety and efficacy or answer questions about its long-term safety or performance.
How much Clinical Data is enough? The Quality and Quantity debate
The EU MDR emphasises clinical evidence quantity and quality as sufficient clinical data. With more notified body feedback, expectations are becoming clearer. Clinical data must meet MDCG evidence level, characteristics, design, statistical power, sample size, follow-up demographics, etc. Scientific contribution should be considered with clinical evidence.
To justify the ‘Intended Purpose’ and prove the device’s claims, manufacturers must compare the total amount of data from pre-clinical studies, literature and SoTA sources to ascertain if there is sufficient clinical evidence. This corresponds to the number of datasets and patients enrolled. The data should include the intended use, indications, clinical claims, contraindications, target groups, and safety and performance objectives based on the State of the Art. They should all be based on the manufacturer’s device and any equivalent device if applicable. A pre-specified method evaluates the identified data sets.
We must quality check identified data sets to meet General Safety and Performance Requirements and claims. PMCF scientific surveys can actively collect clinical evidence. This data updates risk analysis and clinical evaluation. Surveys during PMCF take less time than randomised or single-arm clinical investigations and registry studies.
Investment in Clinical Investigations – what is at stake?
Sufficient clinical data does not mean all medical devices must do expensive ‘Randomised Control Trials’. This is not a pre-requisite for Class III or implanted devices. Consider MDCG 2020-6 (see sections below), rank 1-4 studies do not prescribe RCT’s. Although RCT’s, if designed well, provide high quality evidence. They are the most expensive trial design types. Prospective randomised designs and difficulty recruiting patients raise investigational costs per patient. Manufacturers should use the CDP to assess clinical data needs.
The goal of pre-market studies is to access the market, but EU MDR also requires post-market clinical data. Hence manufacturers will need to invest in further clinical studies post market. Spending most of the budget on RCT’s might cripple future budgets for PMCF studies which can make the product more profitable as they can support future claims, broaden the ‘Intended Purpose’ and differentiate the product in the market.
A manufacturer starting a project must analyse the devices’ use and provide a detailed State of the Art description that explains the devices clinical benefits, risks, safety/performance endpoints and provide a reference range of the safety/performance effect size. The Notified Body requires a detailed document and the manufacturer needs to benchmark their device with objective evidence. MDR requires substantial evidence to prove the device sits within range of the general safety and performance requirements (GSPR) for the State of the Art.

We must analyse the State of the Art and device technical data to define objectives and study endpoints.
Notified Bodies expect patient-level Scientific Surveys for certain devices. Especially for higher-risk class and long-term use/implantable devices with little clinical evidence.
All risk classifications can be surveyed, although the methods will vary. Manufacturers can’t conduct a simple use-related questionnaire for a IIb class device. This wouldn’t give information on the hierarchy level of the medical device, the patient, or the procedure type. Therefore, it would be an invalid way to collect clinical evidence for higher risk devices.
The process’s statistical rationale for the number of patients/surveys should be based on the study’s endpoints, success, and certainty objectives. Biostatisticians calculate the amount of patients/surveys needed to prove endpoints from several sources and translate CER objectives into endpoints. However, survey data may be relevant but not adequate evidence.
Manufacturers must use the clinical evaluation process to evaluate the product’s stage (time on the market), information gathered, and clinical gaps in the planning phase. The CEP, CDP, and CER must describe the assessment for all devices. Nonetheless, the PMCF plan should incorporate surveys for post-market clinical evidence collection.
MDCG 2020-6
This guidance is very helpful to understand the hierarchy of data and clinical evidence requirements.
The MDR does not define whether a survey, full clinical study, or PMS data is sufficient. The MDR requires clinical evidence to support the devices claims, but the MDCG2020-6 guideline provides useful information. As illustrated in the table below, the guideline hierarchy includes full clinical studies, randomised studies, registry studies, and high-quality surveys.
Suggested hierarchy of clinical evidence for confirmation of conformity with relevant GSPRs under the MDR
Rank | Types of clinical data and evidence | Considerations / comments |
1 | Results of high quality clinical investigations covering all device variants, indications, patient populations, duration of treatment effect, etc | This may not feasible or necessary for certain well-established devices with broad indications (eg Class IIb legacy sutures, which could be used in every conceivable patient population) |
2 | Results of high quality clinical investigations with some gaps | Gaps must be justified / addressed with other evidence in line with an appropriate risk assessment, and clinical safety, performance, benefit and device claims. Assuming the gaps can be justified, there should be an appropriate PMCF plan to address residual risks. Otherwise, manufacturers shall narrow the intended purpose of the device until sufficient clinical data has also been generated. |
3 | Outcomes from high quality clinical data collection systems such as registries | Is there sufficient evidence of the quality of the data collected by the registry? Are the devices adequately represented? Is the data appropriately stratified? Are the endpoints appropriate to the safety, performances and endpoints identified in the clinical evaluation plan? |
4 | Outcomes from studies with potential methodological flaws but where data can still be quantified and acceptability justified | Many literature sources fall into this category, due to limitations such as missing information, publication bias, time lag bias, etc. This applies equally to publications in the peer-reviewed scientific literature. However, for legacy devices |
Class III legacy devices and implantable legacy devices which are not well-established technologies should have sufficient clinical data as a minimum at level 4. Those devices which are well-established technologies may be able to confirm conformity with the relevant GSPRs via an evaluation of cumulative evidence from additional sources as listed below. Reliance solely on complaints and vigilance is not sufficient. | ||
5 | Equivalence data (reliable / quantifiable) | Equivalence must meet MDR criteria. It is normally expected that manufacturers should gather data on their own devices in the post-market phase, therefore reliance on equivalence should be duly justified, and linked to appropriate PMCF or proactive PMS. |
6 | Evaluation of state of the art, including evaluation of clinical data from similar devices as defined in Section 1.2 of this document | This is not considered clinical data under the MDR, but for well-established technologies can only be considered supportive of confirmation of conformity to the relevant GSPRs. Data from similar devices may be also important to establish whether the device under evaluation and similar devices belong to the group of devices considered as “well established technologies” (WET). See section 1.2 in this document for the criteria for WET. Data from similar devices may be used, for example, to demonstrate ubiquity of design, lack of novelty, known safety and performance profile of a generic group of devices, etc. |
7 | Complaints and vigilance data; curated data | This falls within the definition of clinical data under MDR Article 2(48) but is not generally considered a high quality source of data due to limitations in reporting. It may be useful for identifying safety trends or performance issues. High volume data collected within a robust quality system may provide supportive evidence of device safety. |
8 | Proactive PMS data, such as that derived from surveys | This falls within the definition of clinical data under MDR Article 2(48), but is not generally considered a high quality source of data due limitations associated with sources of bias and quality of data collection. It may be useful for |
9 | Individual case reports on the subject device | This falls within the definition of clinical data under MDR Article 2(48), but is not considered a high quality source of data due to limitations in generalising findings to a wider patient population, reporting bias, etc. It may provide supportive or illustrative information with respect to specific claims. |
10 | Compliance to non-clinical elements of common specifications considered relevant to device safety and performance | Common specifications which directly address clinical investigation or data requirements would rank higher in this hierarchy. Common specifications may address clinically relevant endpoints through non-clinical evidence such as mechanical testing for strength and endurance, biological safety, usability, etc. |
11 | Simulated use / animal / cadaveric testing involving healthcare professionals or other end users | This is not clinical data but may be considered evidence of confirmation of conformity to relevant GSPRs, particularly in terms of usability, such as for accessories or instruments. |
12 | Pre-clinical and bench testing / compliance to standards | Pre-clinical and bench testing may address clinically relevant endpoints through non-clinical evidence such as mechanical testing for strength and endurance, biological safety, usability, etc. |
Manufacturers must assess their device’s class to determine sufficient evidence.
The manufacturer may combine clinical evidence collection methods depending on the device class. Manufacturers should use the MDCG guideline to establish which approach is sufficient for the device’s claim or objective.
ISO 14155:2020
ISO 14155 provides guidance and requirements for the design, conduct, recording and reporting of clinical investigations. This is in accordance with the MDR and the ethical principles set out in the Declaration of Helsinki.
The manufacturer must consider the applicable part of the ISO 14155 standard. By doing so, the manufacturer understands what data is needed for surveys and the final report. The manufacturer should describe if they met the proposed endpoints and their data findings. This data is included in the CER and PMCF.

Benefits and Claims
In accordance with the MDR, the performance of a device is its ability to achieve its intended purpose as stated by the manufacturer. By extension, the clinical performance of a medical device is the ability of the device to achieve its intended purpose, thereby leading to a clinical benefit when used as intended. ‘Clinical benefit’ means the positive impact of a device on the health of an individual. This is expressed in terms of a meaningful, measurable, patient-relevant clinical outcome(s), including outcome(s) related to diagnosis, or a positive impact on patient management or public health.
Claims can be both clinical and technical. Technical claims may pertain to the design of the device, aka it is reusable, sterile. Whereas any clinical claim but be measurable in-patient outcomes. The notified authority or the national marketing claims regulator will not accept it if you infer a clinical benefit. For instance you cannot say “our device is sold sterile and hence prevents infections” unless you can prove in a study that the rates of infections is lower in your device versus a competitor that is not sold sterile. But it may be that the state of the art requires all devices in your device category to be sold sterile, so it’s better to claim in the intended use that the device is sold sterile.
Notified Bodies state that medical device safety and performance claims, as well as clinical benefit and claims are relevant. Benefits must be measurable, patient-oriented, and meaningful. When the device indirectly benefits a procedure, it should be mentioned. Nonetheless, manufacturers must limit their claims and identify mandatory from complementary information as clinical evidence must substantiate the device’s benefits.

Clin-r+ recommendations
They gave notified bodies the authority to define state-of-the-art in accordance with their expertise and understanding of the law.
Where the manufacturer provides no or an ill-defined SoTA, it opens the door for the Notified Body to impose their interpretation of the SoTA. Uncertainty may need costly clinical trials or no CE mark.
Defining the ‘state of the art’ should be the starting point in the development process by any manufacturer or innovator in MedTech.
If a manufacturer cannot show the Notified Body a thorough understanding of the SoTA for the device, it will leave various areas of the Technical Document susceptible to scrutiny and create uncertainty whether conformance is achieved.
A state of the art literature review gives a wealth of information.
It overviews the performance and safety data, as well as defining the technology’s clinical benefits, disadvantages, risks, and limitations. It contains powerful strategic insight on if a clinical investigation is needed, what type of design would be best to address your gaps, help support your ‘Intended Purpose’ and help keep your trial budget in check.
Manufacturers without this expertise should consider partnering with an experienced consultancy when assessing standards, risk, clinical data and considering what clinical studies are best suited for your devices CE marking needs. Consultancies also come with resources such as clinical study designers, statisticians and experienced SoTA medical writers systematic review helping expand your companies’ capabilities cost-effectively.
Should you have any questions or need professional assistance, CLIN-r+ has a wealth of experience in clinical investigations and literature searches to call upon. Get in touch!
