Clinical Development Plan (CDP)
EU MDR compliance & strategic planning of Medical Devices
The EU MDR Technical Documents (TD) now requires a new document known as the Clinical Development Plan (CDP). It is a valuable strategic document that can assist MedTech in aligning its development and regulatory requirements.
A Clinical Development Plan is required by section 1(a), eighth indent, of Annex XIV of the Medical Devices Regulation (EU) 2017/745 (MDR). Annex XIV, section A of the MDR contains a definition of the CDP. A Clinical Development Plan is a strategy that shows progression from preliminary research, such as first-in-man studies, feasibility research, and pilot studies, to conclusive research, such as pivotal clinical investigations and a PMCF, with an indication of milestones and a description of prospective acceptance criteria. The European Medical Device Coordinating Group’s (MDCG) MDCG 2020-1 Guidance on Clinical Evaluation (MDR) / Performance Evaluation (IVDR) provides additional help for this. The purpose of this guidance is to offer direction for medical device software. Insights about planning and the necessity of a Clinical Development Plan, for example, are contained that are relevant to various medical devices.
This paper aims to clarify the proper organisation of a CDP. It’s a useful early tool to make regulatory submissions easier while manufacturers are still developing their product (s).

Suggested document structure for CDP
A sample CDP is shown in the table over the page. The initial Business Development rationale is interpreted into the clinical usage parameters by the CDP, which is a key strategic document. It accomplishes this by outlining the medical device’s Intended Use, Intended Purpose, Patient Population, Intended User, Intended Environment, and the key operational principles. The CDP then aligns essential business functions by specifying key deliverables required to establish clinical claims, obtain CE marking, and identify important differential design (to obtain clinical and technical claims), all of which are necessary to facilitate the marketing and smooth market entry of the product.
It therefore makes sense that CDP shouldn’t be a stand-alone document, but rather a core one that unifies different business processes and outlines the crucial contributions that each department must make in order to obtain the CE mark, gain access to markets, and keep the device competitive.
Table 1 – Suggested CDP Structure
| Section | Title |
| 1 | Scope of Clinical Development Plan |
| 2 | Clinical Context: Clinical Background, Current Knowledge, State of Art |
| 3 | Device overview |
| 3.1 | Description of Target Device(s) |
| 3.2 | Intended Purpose |
| 3.3 | Indication |
| 3.4 | Intended Patient Population |
| 3.5 | Intended Use, Intended User, Intended environment |
| 3.7 | Safety and Performance Claims |
| 4 | Developmental Context for Device |
| 4.1 | Rationale for the Development |
| 4.1.2 | Maturity of Technology |
| 4.2 | Clinical Need and Underlying Pathology |
| 4.2.1 | Mechanism of Action |
| 4.3 | Design Plan |
| 4.3.1 | Potential Bench Testing Properties, applicable common standards, and Key Outputs |
| 5 | Commercial Rationale for Development |
| 5.1 | Clinical Target(s) and Descriptions of Disease |
| 5.2 | Epidemiologic Considerations |
| 5.3 | (M) Unmet Clinical Need(s) |
| 5.4 | (M) Unmet Market Need(s) |
| 5.5 | (M) Market Assumptions and Projections |
| 5.6 | (M) Product Profiles: Optimal versus Minimal Acceptable |
| 5.7 | (M) Synergy with Other Company Products (Marketed or Under Development) |
| 5.8 | (M) International Considerations |
| 5.9 | (M) Competitive Situation (Existing and Potential) |
| 6 | Market Access |
| 6.1 | Country Footprints and Jurisdictions |
| 7 | Clinical Data – Study Plan |
| 7.1 | Clinical Data Needs |
| 7.2 | Pre-Market |
| 7.3 | Post-Market Surveillance |
| 7.3.1 | Post-Market Clinical Follow-up |
| 8 | Regulatory Considerations |
| 8.1 | Regulatory Submissions Planning |
| 8.1.1 | Conformity route per jurisdiction |
| 8.2 | Clinical Investigation Documentation |
| 8.4 | Regulatory Trends and Intelligence |
| 8.5 | International Regulatory Planning and internal harmonisation |
| 9 | Overall Strategic Planning |
| 9.1 | Product Lifecycle Planning |
CDP for alignment of terminology
Terminology is crucial in regulatory contexts and frequently contributes to audit conclusions. Snags are encountered when transitioning from US, Australian, or EU MDD. By utilising information from your pre-existing CE marks, effective translation increases the efficiency of gaining markets. Early completion of this task allows your team to concentrate on the gaps that must be filled in order to comply with the GSPR.
CDP for new versus existing medical devices
With regards to any new devices where clinical evidence is still to be compiled, consideration must be dedicated to outline how pre-clinical data, first-in-man studies, feasibility, pilot, and pivotal studies can proceed to create the evidence needed to apply for the CE mark.
The content of the CDP is less clear for devices that are already in use. The procedures outlined above should have been followed, and the outcomes should have been covered in the CER, as these devices are already certified. The majority of CDP requirements do not apply to legacy devices, with the exception of the post-market clinical follow-up (PMCF) plan, according to MDCG guidance 2020-6 on the clinical evidence needed for medical devices previously CE marked under Directives 93/42/EEC or 90/385/EEC. Milestones and acceptance criteria should be mentioned in the PMCF plan.
Life cycle management of clinical/performance data in clinical evaluation (MDR) / performance evaluation (IVDR) process
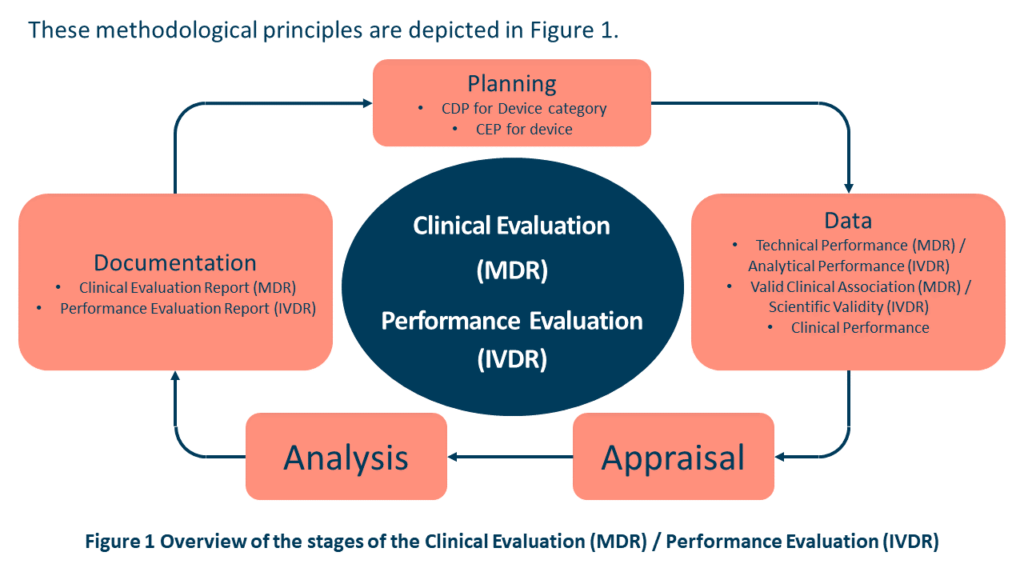
Over the course of the device’s lifespan, the manufacturer should actively and consistently monitor the device’s performance, effectiveness, and safety. The manufacturer must also specify how future product releases or claims will be handled to guarantee that the required clinical data is available to support the claims and their intended use. Such data should be subject to the Clinical Evaluation (MDR) / Performance Evaluation (IVDR) principles shown in Figure 1 from MDCG 2020-9 Figure 1 and Annex 1. Post-market information such as complaints, PMCF/PMPF data, REAL-WORLD PERFORMANCE data, direct end-user feedback, or newly published research / guidelines are just a few examples of post-market information that may be included.
Access to Real-World Performance data is made possible by the MDSW’s exceptional level of connectivity and can be utilised for a variety of things, including but not limited to:
- timely detection and correction of malfunctions.
- detection of systematic misuse.
- understanding user interactions.
- to conduct ongoing monitoring of Clinical Performance.
- to improve effectiveness.
- develop the claims in the Clinical Development Plan (MDR) or future releases.
Devices for which CDP obligations are less clear
The CDP is still required for devices that have already received CE marking under the MDR as well as for those that have already received CE marking under the MDR because it contains crucial PMS data gathering insights like the justification of the PMCF plan and potential future “Indications” that the manufacturer may work toward.
CLIN-r+ recommendations
In light of this, CLIN-r+ suggests that manufacturers create their CDPs, summarising the history of clinical evidence, citing reports, outlining the current state of the art, in addition to any ongoing activities.
The CER can be used to supplement such evidence and weigh the clinical advantages and disadvantages. The summary history component of the CDP will expand over time whilst the rest narrows down to just PMCF or future indications. In order to facilitate future filings, the CDP will give the Notified Body reviewer a guide of how the clinical data for a device has been put together as well as the manufacturer’s long-term goals.
CLIN-r+ offers a full clinical development service, as well as templates for any section of the Technical Documentation required, including the CDP. Our templates have been completely updated to reflect the most recent requirements. Get in touch!
