Post Market Clinical Follow up (PMCF)
EU MDR Compliance
The European Medical Devices Regulation (EU MDR) impacts medical device manufacturers with plans to get devices approved in the EU as well as manufacturers with existing products on the market. This means that going forward, the requirements under MDR will be much more stringent.
The EU MDR will increase the need for Post Market Surveillance (PMS), and therefore Post-Market Clinical Follow-up (PMCF). This also changes the requirements for PMCF data. Manufacturers will need to collect more clinical evidence and improve data quality and management for each device.
What is Post Market Clinical Follow up (PMCF)?
MDR Annex XIV(B, defines Post-Market Clinical Follow-up (PMCF) as:
‘A continuous process that updates the clinical evaluation and that shall be addressed in the manufacturer’s post-market surveillance (PMS) plan.’
PMCF is a systematic collection of clinical data, documentation, and evidence to proactively identify safety or performance issues in a CE-marked medical device and update its clinical evaluation. PMCF is a crucial part of medical device post-market surveillance, supplementing existing pre-market clinical and non-clinical data. The Clinical Evaluation Report (CER) and technical documentation must include a PMCF evaluation report. Medical devices should have continual PMCF activity throughout their lifetime.
Objectives for PMCF under the EU MDR include:
- Identifying and investigating residual risks associated with use of the device.
- Contributing towards the update of Clinical Evaluation.
- Detecting new risks and side-effects.
- Confirming the devices overall safety and performance.
- Identifying systematic device misuse and the impact on safety and performance.
Manufacturers must document PMCF activities in a Post-Market Clinical Follow-up Plan. Then we must collect the PMCF results in a PMCF Evaluation Report, which also forms part of the Clinical Evaluation Report (CER).
Ensuring your PMCF Plan is EU MDR compliant
EU MDR PMCF requirements will increase medical device manufacturers’ PMCF expenditure. As PMCF processes become proactive and end-user transparency of device quality increases, manufacturers of high-quality medical devices may gain substantial advantages. The greatly increased amount and quality of data acquired for each device will give manufacturers valuable insights into their device performance. We can utilise this to improve marketing or design future medical devices faster and more efficiently. To take advantage of these benefits, manufacturers must gather and manage PMCF data methodically and comprehensively.
EU MDR Annex XIV Part B states that the PMCF plan must at least include the following:
- The general methods and procedures of the post-market clinical follow-up to be applied.
- The specific methods and procedures of PMCF to be applied, such as evaluation of suitable registers or clinical studies.
- A rationale for the appropriateness of the methods and procedures referred to in points (a) and (b).
- A reference to the relevant parts of the clinical evaluation report referred to in Section 4 and to the risk management referred to in Section 3 of Annex I.
- The PMCF will address the specific objectives and clinical data gaps (provided by the CER or from Clinical Affairs to outline the post-market clinical data needs).
- An evaluation of the clinical data relating to equivalent or similar devices (obtained by the State-of-the-Art literature review).
- Reference to any relevant CS (common specifications), harmonised standards when used by the manufacturer, and relevant guidance on PMCF.
- A detailed and adequately justified time schedule for PMCF activities (e.g. analysis of PMCF data and reporting) to be undertaken by the manufacturer.
Manufacturers should have clearly defined clinical data gaps and complete the checklist from MEDDEV 2.12-2 when a PMCF study is needed.
A State of the Art (SoTA) literature review for the device provides key insights in safety and performance endpoints, as well as clinical data from the SoTA. It’s also important to collaborate with Clinical Affairs, regulatory and engineering to pinpoint gaps in conforming to the GSPR. This will ensure important cross functional alignment. You also need a PMCF plan template that is not only compliant and contains the correct amount of information, but also reusable across products and markets. We’d recommend the template provided by the MDCG work group to ensure alignment. As getting your template right from the start is critical to success.
We share more key insights on PMCF planning in our white paper ‘Three Tips for PMCF Planning’.

Selecting the Appropriate PMCF Activity
The PMCF plan must describe the methods applied to gather data on clinical performance and safety. Due to a lack of guidance, some medical device manufacturers have struggled to choose a PMCF approach under the EU MDR. The MDR requires the PMCF strategy to incorporate scientifically justified methods, but MDCG has not provided practical instructions on how to do this.
The most commonly used PMCF data gathering methods are:
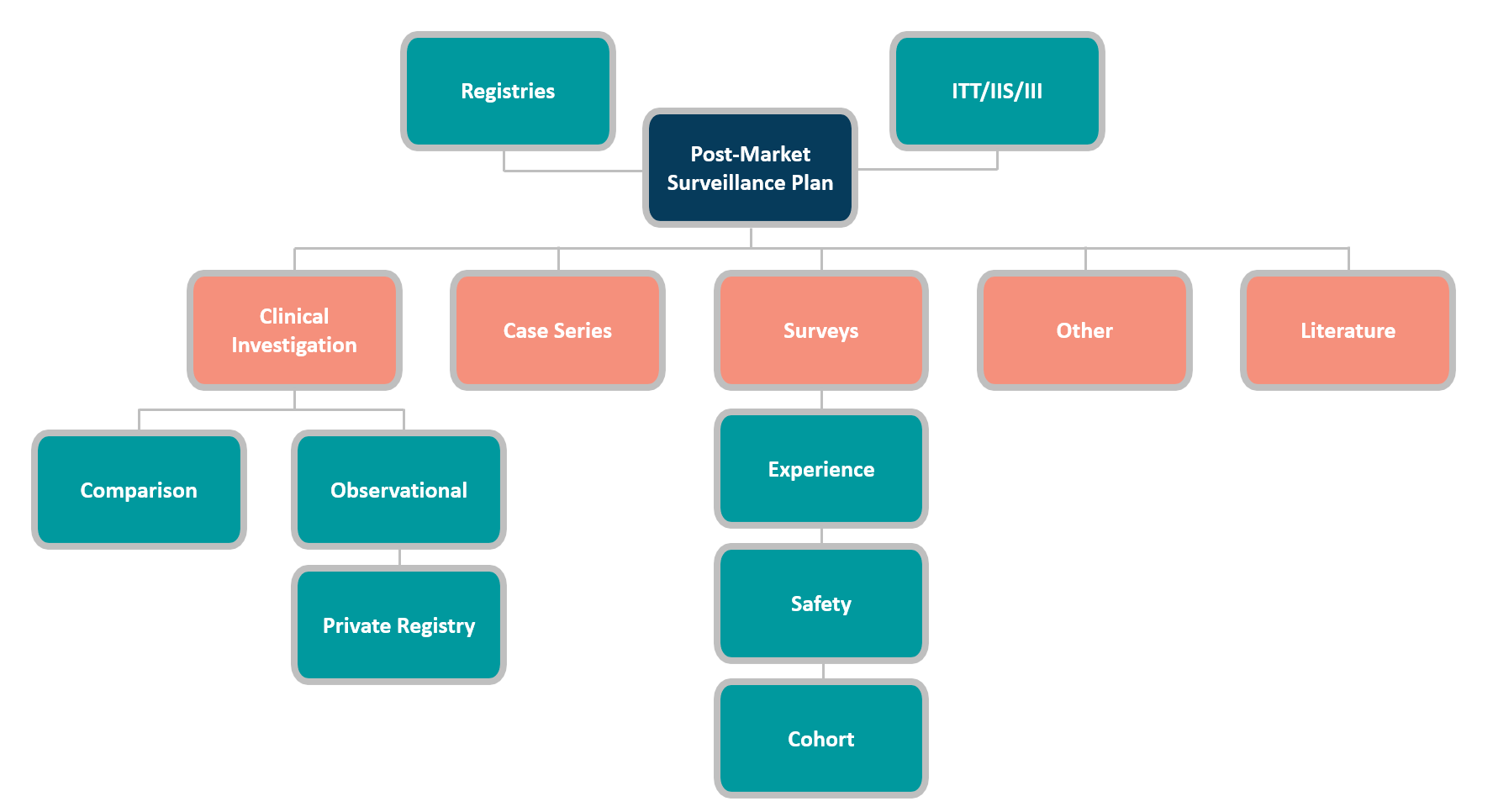
Each PMCF approach has pros and cons, and its usability depends on several aspects. Before choosing a PMCF activity, carefully consider these factors. The prominent deciding factors are:
- Medical Device classification and PMS reporting requirements.
- Sales numbers and markets.
- Data available from earlier Clinical Evaluations (gap analysis).
- Device performance measure and clinical/industry standards.
- Whether you have access to customers.
- Whether you have access to end-users.
- Whether you have access to the patients.
- How the device is used/applied in practice.
Whether there’s a data consent in place (from existing data sources).
Post Market Clinical Follow up (PMCF) Survey
Manufacturers can collect post-market data via PMCF surveys. The EU MDR has changed PMCF survey requirements, along with most areas of Post-Market Surveillance (PMS). The sponsor must use best practice data collection procedures to conduct a PMCF survey and satisfy Notified Bodies. There are four key areas to consider:
- What is the appropriate PMCF sample size?
- Questionnaires need to provide reliable and usable data.
- Use a validated tool to simplify Good Clinical Practice (GCP) compliance.
- Choose a tool with several ways to initiate surveys.
A PMCF survey is cheaper than a clinical examination, but we must do it correctly to avoid delays, extra expenditures, or loss of CE-marking. Manufacturers should avoid using non-validated data gathering tools, as Notified Bodies can claim that the data collection does not meet ISO 14155 regulations on electronic data capture, which can leave the data collected unusable.
Sample Size for Clinical Investigations and PMCF Studies
Medical device sample size calculation is complicated. Medical device sample size calculations typically use five inputs. You must define or make assumptions on all of these inputs. Values vary on the study objective, endpoints, and design, and the sample size calculation is susceptible to the value inputs used, making it prone to be imprecise. The five basic sample size calculation inputs are:
- Statistical Hypothesis.
- Significance level.
- The minimal clinically meaningful effect.
In addition, these factors should also be taken into account:
- Test Margin
- Subject Drop-out rate
- Treatment allocation
Sample size calculation is important, and we recommend utilising external expertise for guidance when possible.
Adverse Event Reporting Under MDR
The MDR requires Good Clinical Practice (GCP) compliance for both pre- and post-market clinical investigations. A successful clinical trial requires recording and reporting adverse events (AE) and serious adverse events (SAE) and keeping up with regulatory changes.
It’s important to note that there are differences in reporting adverse events in Clinical Investigations and PMCF Investigations under the EU MDR. However, it depends if we carry the PMCF investigation out on a CE marked device under its intended use or not. If a device is CE marked and the investigation is observing outcomes under its intended use, MDR Article 80(5) and 80(6) govern the requirements for safety reporting. Therefore, the guidance on vigilance in Articles 87 to 90 of the MDR applies instead of Article 80(1) to 80(4).
Manufacturers are only required to notify competent authorities of serious incidents that have not been thoroughly documented (known side-effects explicitly indicated in the product documentation) before launching the device to an EU market. When used in the EU, manufacturers are also required to report safety corrective actions on devices used in third countries.
Clinical investigation reports must include all adverse events. Thus, pre- or post-market investigations have the same documentation requirements. However, post-market clinical investigations may require adverse event reporting under national legislation. Manufacturers must recognise each country’s specific requirements.
Real World Data (RWD) and Real World Evidence (RWE)
RWD and RWE are playing an increasing role in health care decisions and the FDA currently uses both of these to monitor post market safety and adverse events to make regulatory decisions. This data supports coverage decisions, develops guidelines and supports tools for clinical practice.
Medical device developers can use RWD and RWE to support clinical trial designs, such as large simple trials and pragmatic clinical trials, along with observational studies, to explore new treatment methods.
Real World Data relates to patient health status and/or delivery of healthcare routinely collected from various sources. As an example, these sources include:
- Electronic Health or Medical Records (EHRs/EMRs).
- Product and disease registries.
- Data from wearable devices.
- Data from mobile devices.
- Patient reported outcomes (PRO) data.
- Specialised/secure websites or tablets.
In order to use RWD to generate evidence, the data must be of sufficient quality and fit for use in medical device regulatory decision making. Studies must meet regulatory requirements.
We can collect rWD throughout a medical device’s life cycle, from pre-market clinical investigations (CIs) to post-market surveillance (PMS) of real-world usage. It is used to verify a device’s safety and performance before market release, and provide adequate post-market surveillance.
Real World Evidence (RWE) is clinical evidence from RWD analysis of a medical product’s use and potential benefits or risks. Various studies or analysis can generate RWE, including:
- Randomised trials.
- Large sample trials.
- Pragmatic trials.
- Observational studies.
We can use RWE when clinical trials cannot account for the entire patient population of a particular disease. Patients with comorbidities or a distant geographic region or age limit who did not participate in any clinical trial may not respond as expected to the treatment. RWE solves these problems.
Clin-r+ recommendations
Start your PMCF journey by having suitable processes and templates formulated to ensure all required information is formulated and adequate. With your first PMCF plans, draw and test the process with a few representative and high-priority devices. Then pilot and refine it from the PMCF plan draft to the completion of the PMCF report. This will be used to test templates, forms, and processes to ensure that they perform well across your organisation. It’s also vital to remember that the PMCF process isn’t a one-and-done affair. Manufacturers must consider how the PMCF process interacts with the PMS, clinical evaluation, and risk management processes, and how the papers will be updated.
Preparing for MDR and IVDR in a timely manner (even early, if possible) requires a balance of in-house and outsourced expertise. Medical device manufacturers can build and implement successful PMCF roadmaps by partnering with third-party specialists.
To learn more about CLIN-r+ post market services for medical devices and IVD manufacturers, get in touch!
