

Timing is crucial when implementing a QMS. The ideal time is when enough information and knowledge have been gathered to ensure an efficient QMS development process. This is usually around the design and development phase.
However, improvements may be necessary to existing QMS processes. If you already have a QMS in existence, it may be necessary to refine or update it to ensure you are in line with all the requirements. Some of the key processes are outlined below.
Medical Device File
Every medical device type or device family must have a technical file (MDD) or technical documentation (MDR). The expected contents of the file are listed in the regulation and for EU MDR compliance, manufacturers should familiarise themselves with Annex 2 and 3 of the MDR which outlines requirements.
Document Control
Document control is the process for managing all the different documents that circulate throughout the lifecycle of the device. Documentation is necessary for every aspect of a QMS. The document control procedure should define your criteria for document control. This includes version control along with document review, approval processes, change history and storage.
Record Control
Managing records and managing documents are two different things but are sometimes confused with each other. Document control is outlined above. However, records are evidence that certain processes have been followed. The same criteria are applicable regarding review and approval as it is for document control, but records do not typically have versions.
For example, a record would demonstrate the device realisation process and that the device meets defined requirements. Other record examples are servicing or sterilisation. If the device requires either of these then records must be kept to document this.
Design Control
The purpose of design control is to control the entire design process and ensure the devices’ design meets user needs, intended use(s) and specified requirements. There are various elements manufacturers must account for as shown below.
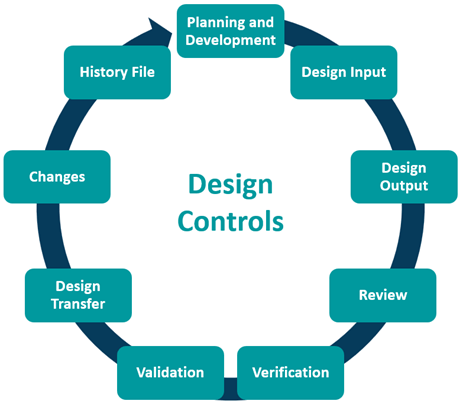
Management Responsibility
Top Management are integral to QMS. They are responsible for several stages and overseeing the QMS. Other areas they must be involved in include quality policy, quality objectives, communication and review at planned and recorded intervals.
Training
Manufacturers must have specifically trained and competent employees in place. Therefore, a record of all qualifications and training must be kept and maintained. Companies may need to provide training and assess effectiveness to ensure their employees have the required skills to perform their duties.
Audits
A proper audit process must be in place with regular quality audits being conducted. This is to ensure that the whole QMS is compliant. Manufacturers need to fully audit all applicable requirements to ensure compliance. Where any audit findings occur, this must become a non-conformance and be recorded as such.
The best practice for managing a QMS is to continually monitor its effectiveness and ensure any necessary adjustments are made.
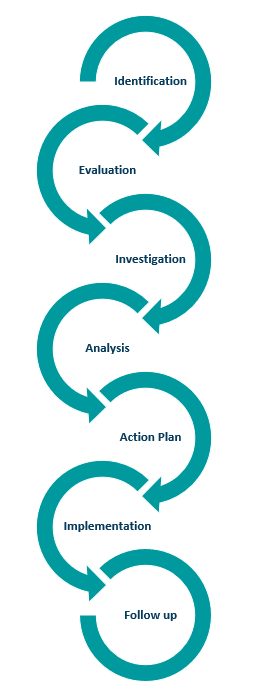
Corrective and Preventative Actions (CAPA)
A CAPA process must be implemented.
The purpose of CAPA is to standardize the response when corrective action is required.
This process is essential for manufacturers to resolve and prevent issues which are likely to impact the device quality.
This can mean collecting information/data, identifying, and investigating device and quality problems, and effecting corrective and preventive action.
It also encompasses verifying or validating corrective and preventive actions, communicating corrective and preventive actions as appropriate, and providing information for management review.
All these activities must be documented.
There are several steps in a CAPA process as shown here.
Production Controls
The purpose here is to ensure you are manufacturing products that meet specifications, develop adequate processes, validate them, and then monitor and control the entire manufacturing process.
Manufacturers must monitor production processes to ensure that device manufacturer is continuously planned, executed, monitored, and controlled. Typically, this is to ensure that the device meets defined specifications and the necessary processes and correct environment for this are in place.
