Documentation requirements in 2023 for compliance with the new ISO revisions
When it comes to medical devices, regulatory standards are subject to change. As technology changes, new data becomes available, and old ways of looking at things become outdated, the updated guidance becomes the new conformity expectation. The Toxicological Risk Assessment and Biological Evaluation documentation will undergo significant modifications with the release of the much awaited revisions in this area.
In this whitepaper, we highlight the changes made in 2023 to toxicology and biological evaluation that manufacturers need to be aware of and what it means for the documentation required to be compliant.

What do the revisions mean?
The revision of ISO 10993-17:2002 “Biological Evaluation of Medical Devices — Part 17: Establishment of Allowable Limits for Leachable Substances,” has been released.
The new version, ISO 10993-17:2023, is now called “Biological Evaluation of Medical Devices — Part 17: Toxicological Risk Assessment of Medical Device Constituents.” The revised ISO aligns with current best practices in toxicology and other ISO 10993 standards. It also introduces new tools to evaluate chemical constituents. This means that toxicologists are able to evaluate these more consistently. The standard also introduces new important concepts and provides guidance on the structure for toxicological risk assessments and the methodology for material. ISO10993-18 had a seven-year revision process that resulted in an update in January 2020. Shortly thereafter, ISO 10993-17 was updated using these insights to outline the TRA requirements leading to another amendment being needed to ISO10993-18:2020/AMD1:2023.
As ISO 10993-1 now requires chemical characterization for all device types. ISO 10993-18:2020 helps to clarify why:
- Analytical testing is not always required for chemical characterization.
- There are various possible approaches to the process.
- A lot of work is driven by multiple solvents, extractions, injections, and methods.
Unfortunately, many uncertainties remain regarding:
- How to apply reference standards.
- The reliability of identification.
- The selection of dose based thresholds.
As a result of these changes, the documentation needed to extend the lab tests are now formal. Now, manufacturers have to complete a Biocompatibility Evaluation Plan (BEP), compile a Toxicological Risk Assessment (TRA), and write up all their results in a Biocompatibility Evaluation Report (BER) with a toxicology team.
Biological Evaluation
The overall biological evaluation workflow is pictured below, but we’ll look into each element in more detail.
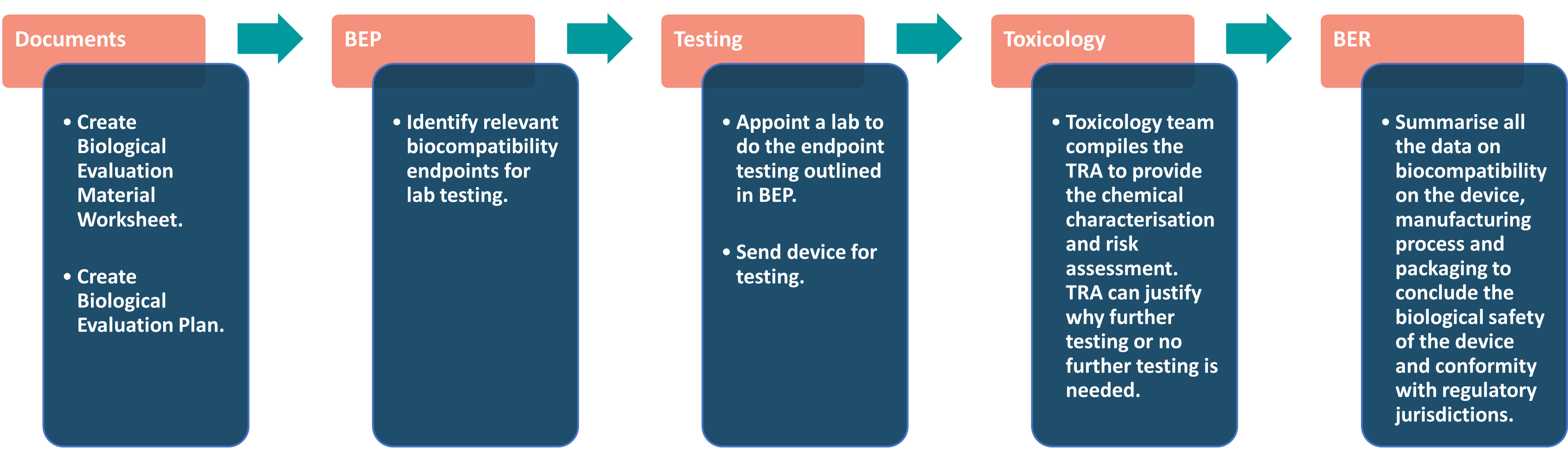
Factors considered during Biological Evaluation:
- The construction material(s) (e.g., all direct and indirect tissue contacting materials).
- Medical device configuration (e.g., size, geometry, surface properties).
- Intended additives, process contaminants and residues during device manufacture.
- The manufacturing process(es).
- Packaging materials that contact the medical device (directly or indirectly).
- Leachable substances & degradation products
- Other components and their interaction in the final device.
- The performance and characteristics of the final device.
- The physical and chemical characteristics of the device.
- Systemic effects and local effects of implantable devices.
- Biological hazards of every material and final product.
- Existing toxicology and other biological safety data.
Biological Evaluation Worksheet
The Biological Evaluation worksheet details the medical device’s configuration, material composition and manufacturing. Using the bill of materials (BOM) it provides a comprehensive list of everything needed to manufacture the medical device. The raw materials, traceability validations, manufacturing process, reprocessing processes and packaging are outlined for potential biological risks it may introduce to the intended user and use patient population. It also specifies the content type and contact time per component.
Biological Evaluation Plan (BEP)
The Biological Evaluation Plan details the medical device strategy. This must address biological risk or biocompatibility for the device. Medical devices require a BEP to examine their configuration, material composition, manufacturing, intended use, any extant testing information and clinical history. The BEP needs to identify any biocompatibility gaps that exist for the device. Expert recommendations can then be provided regarding how to best fill these gaps based on ISO 10993-1 and product or country specific documentation.
BEP development is consistent with current regulatory expectations. ISO 10993-1 also states “The biological evaluation shall be planned, carried out, and documented by knowledgeable and experienced professionals,” which highlights the necessity to involve the appropriate expertise.
Construction Materials – Chemical Characterisation of the Medical Device
Supplier data and raw material data helps determine chemical composition and potential other sub-suppliers that could add the biological risk. Composition can also alter during production processes or storage as the device or materials come into contact chemical substances. These include processing aids, cleaning fluids, mould release agents, process contaminants and sterilisation residues. Materials can also degrade.
Biocompatibility ISO 10993 characterisation studies should identify and quantify medical device materials and their chemical constituents (material composition) in each material of construction. Particular focus should be on screening and determining CMR substances.
Toxicological Risk Assessment (TRA)
All marketed products need toxicity risk assessment and ISO 10993 Parts 17 and 18 guide the risk assessment methodology.
Transient or short-term contact devices may have low danger levels based on available information and component supplier data.
Drug delivery systems must follow ICH, PRQI, and FDA guidelines.
The TRA is conducted using allowable limits, Safety Concern Thresholds (SCT, and Analytical Evaluation Thresholds (AET). After reviewing chemical and material data, the TRA can be submitted to authorities.
Health Risk Assessments (HRA) of Individual Substances
Any existing data, both clinical and nonclinical as well as human exposure data, along with any experience relevant to the device is evaluated. The results are included in the HRAs for individual substances.
The HRA outlines the expected presentation in patients. This gives manufacturers the adverse events that need to be monitored in the PSUR/PMSR.
Biological Evaluation Report (BER
The BER is used to draw the conclusion of the medical devices biological safety for patients and intended users during the course of its use.
It includes various information such as:
- Summaries of all biological tests performed.
- Justifications for any tests not performed.
- Supporting literature data, data evaluation, and gap analysis for biological safety information.
- Rationale for why additional information isn’t required.
- The Toxicological Risk Assessment (TRA) conclusion.
- Summary of all the lab test reports.
A statement verifying the completed biological risk analysis and risk controls.
Clin-r+ recommendations
The Biological Evaluation Plan should be completed prior to conducting extensive biocompatibility testing. However, CLIN-r+ has the expertise to formulate a plan at any stage and include all relevant testing information. We can also provide rationale for the testing strategy according to ISO 10993 and ISO 18562 series standards.
CLIN-r+ can help assure the safety of your medical device materials and components during development, screening, reprocessing, and sterilising. We can also examine your materials’ sustainability to support your ‘net zero’ conversion.
We offer end-to-end Medical Device Biocompatibility Evaluation and Toxicological risk assessment of medical device constituents as per ISO 10993-18 and 17. Working with medical device testing lab experts to provide you with the testing and documentation to help you demonstrate EU MDR and FDA-compliant device material and component safety and biocompatibility.
Should you have any questions or need professional assistance, CLIN-r+ has a wealth of experience to call upon. Get in touch!

Related Articles

Remediating biocompatibility workflows to meet EU MDR compliance requirements
This case study looks at remediating biocompatibility workflows to meet EU MDR compliance requirements. CLIN-r+ supported the manufacturer with their problematic biocompatibility workflows to address …
Read More →

ISO 18562 – Biocompatibility evaluation
ISO 18562, has become the industry standard for testing the biocompatibility of breathing components. It has four parts: general principles, evaluation of particle emission, evaluation …
Read More →

Biocompatibility
Biocompatibility and how to apply extractables and leachables to medical devices. ISO 10993 now requires knowledge of any chemicals released by a device while it …
Read More →