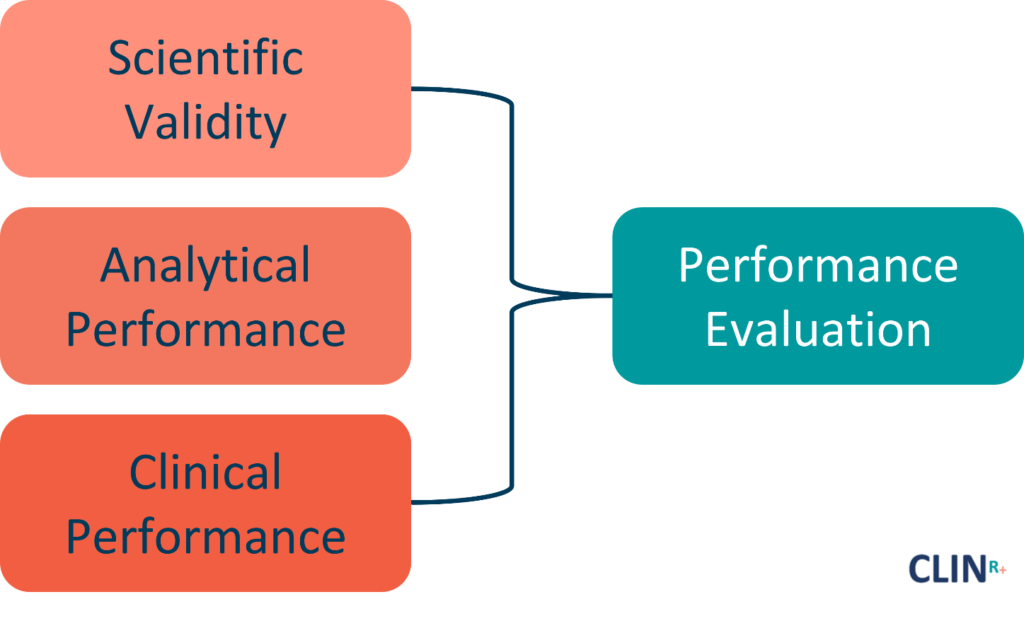
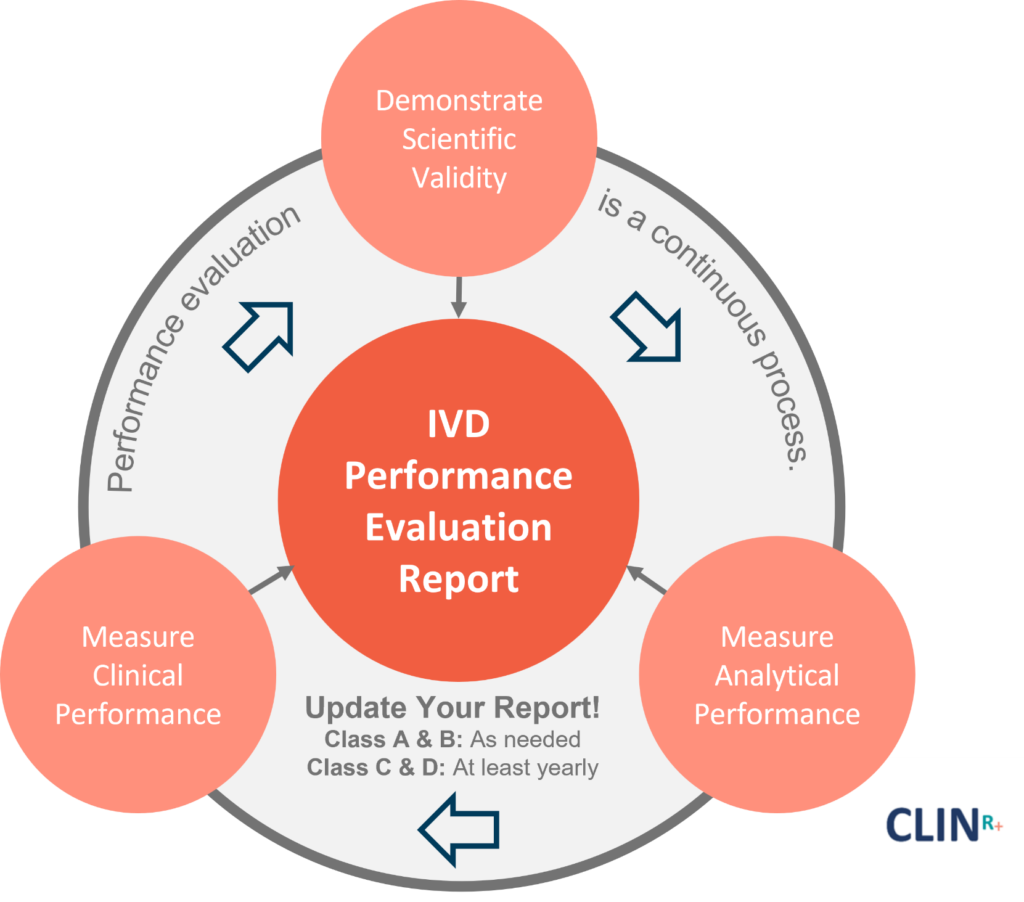
Preventing false positives and false negatives, both of which could lead to detrimental physical and psychological impacts for patients, is among the most important reasons for performance evaluation.
While the temptation to dive into the nitty gritty of the report is there, the writing of your Standard Operating Procedure (SOP) and creating your comprehensive Performance Evaluation Plan (PEP) as part of the planning requirements, are critical to the report’s success. In fact, omitting the SOP would cause immense problems further down the line, since Article 56(3) of the IVDR mandates a process established and implemented.
What if your device already has US FDA 510(k) clearance? Though this may be the case of many, due to the strictness of the EU IVDR regarding the demonstration of clinical benefits, it is highly probable that you will need to generate (and analyse) more data than you have at this moment! Furthermore, the opinions of clinical benefits related to IVD medical devices will vary considerably between the European regulators and US FDA.
 Although no two performance evaluation plans are the same, there are certain elements that should be included in all of them without fail. Including, though not limited to:
Although no two performance evaluation plans are the same, there are certain elements that should be included in all of them without fail. Including, though not limited to:
- Your performance evaluation supports the General Safety and Performance Requirements (GSPR).
- Intended use or purpose of the device.
- Indications for use, limitations, and contra-indications.
- Performance specifications of the IVD.
- A description of the current state of the art.
- Benefit-risk ratio parameters.
- Intended target patient groups.
- Analyte or marker to be used.
- Foundation for underlying software decision making (if applicable).
Dedicate sufficient time to prepare this plan, and it will pay dividends when you complete the performance evaluation, saving you many more hours than those you invested during the planning stage. The possibility of your product’s risk assessment and risk management processes being altered by your “research” is always something to bear in mind. Moreover, it is advisable to consider whether you have the internal resources to administer the numerous aspects of the performance evaluation. Otherwise, bringing in outside expertise may be something that needs consideration.