Risk management appears simple from a distance. You have a device; you assess its potential risks, mitigate those risks, and track them over time. Isn’t it simple? If only it were as simple as that. However, risk management is one of the more difficult aspects of regulatory compliance, simply because risk comes in various flavours and severity perceptions. Furthermore, we can estimate the likelihood of harm occurring in various ways.
The fact that we often don’t have enough real-world data to accurately quantify risks, especially for new devices, makes risk management problematic. Fortunately, you can establish a systematic process for analysing, evaluating, controlling, and monitoring risks. Before we get into that, let’s take a step back and discuss the rules and guidelines that govern how you should approach risk management.
 For medical devices, we define risk and risk management as:
For medical devices, we define risk and risk management as:
- Risk – the combination of probability that harm occurs and the severity of that harm.
- Risk management – the systematic application of management policies, procedures, and practices to the tasks of analysing, controlling, and monitoring risk.
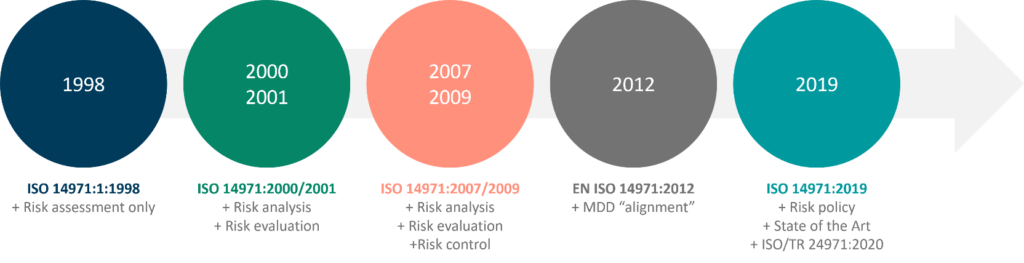
Simply put, we all have a vested interest in ensuring the safety and efficacy of medical devices. Risk management is therefore not optional; it is a legal requirement all over the world. The Quality System Regulation of the United States Food and Drug Administration (FDA) requires it (21 CFR Part 820), and the Medical Device Regulation (MDR 2017/745) in Europe mandates it. Similarly, risk management is required in Japan, Canada, Australia, Brazil, and all other major markets, as stated in their national regulations or ISO 13485:2016.
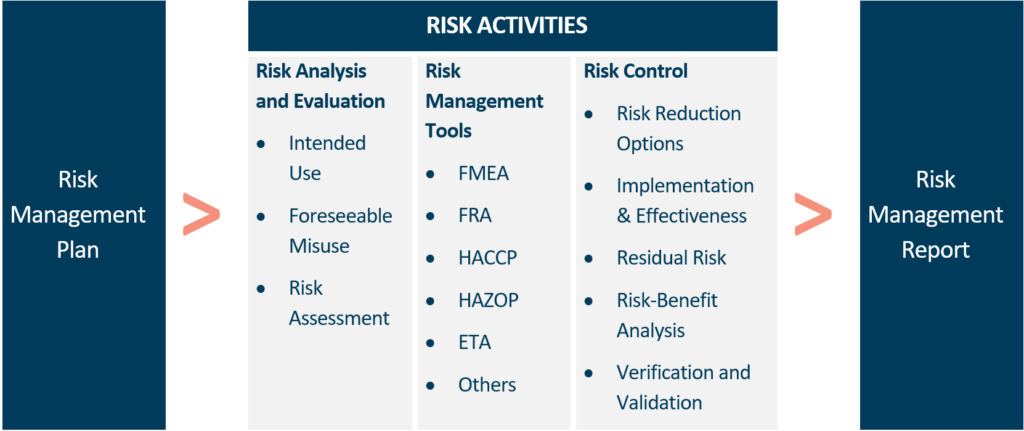
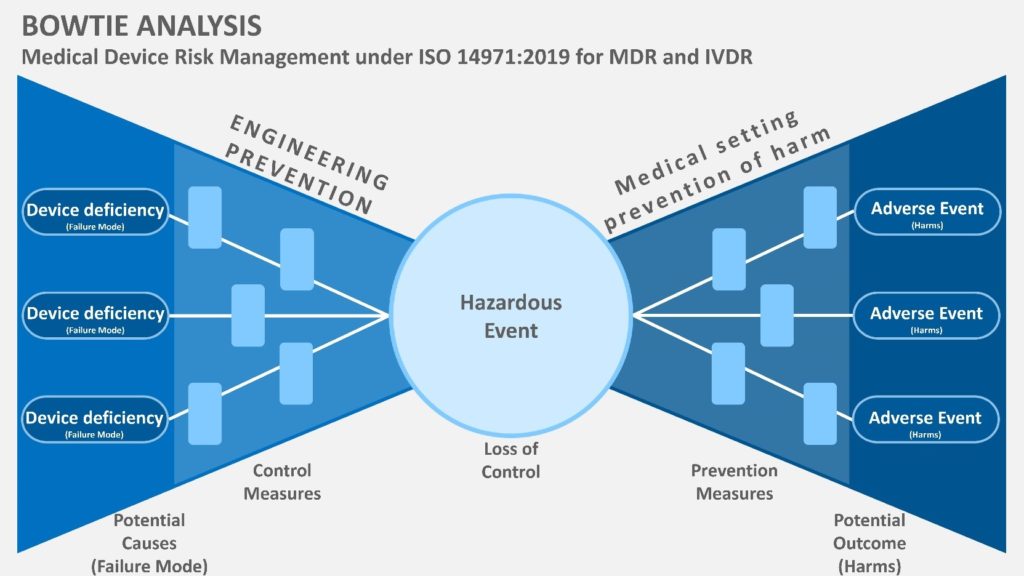
 This is where you give yourself credit for everything you’ve done. Reconnect everything to your original strategy. Did you stick to the plan? Did you keep track of any deviations and justify them? It’s critical that you write clear and simple conclusions, such as “The risk management process outcomes support that the implemented risk control measures reduce the residual risks of my device compared to the clinical benefits.” This goes a long way toward establishing your method’s credibility.
This is where you give yourself credit for everything you’ve done. Reconnect everything to your original strategy. Did you stick to the plan? Did you keep track of any deviations and justify them? It’s critical that you write clear and simple conclusions, such as “The risk management process outcomes support that the implemented risk control measures reduce the residual risks of my device compared to the clinical benefits.” This goes a long way toward establishing your method’s credibility.
It’s a good idea to finalise your plans for risk monitoring throughout the device life cycle at this point. Last but not least, all the documentation you create during these three basic steps becomes the content of your device’s risk management file.