Effective Post Market Surveillance

What your Notified Body wants to see in the Post Market Surveillance Plan and PSUR/PMSR
With the transition to the MDR looming, manufacturers will have to revise their post market surveillance (PMS) plans. The plan must include the proactive gathering of the clinical and safety-related data needed to demonstrate the continued safety of their medical devices with CE marking. This also includes continuous monitoring of performance to properly identify the risks associated with practical, everyday use of the device, and to ensure that there are no uncontrolled risks or security problems.
The MDR defines post-market surveillance as “all activities carried out by the manufacturer… to institute and keep up to date a systematic procedure to proactively collect and review experience gained from devices they place on the market” (Article 2 (60)). This is a significant deviation from the MDD, in which post-market surveillance was considered part of the manufacturer’s obligations to maintain a “systematic procedure to review experience gained from devices in the postproduction phase” (MDD Annex III). PMS was further mentioned in Annex X, but nowhere was it explicitly defined and outlined.
MDR Article 2 clearly defines PMS. It also listed PMS as one of the general obligations of the manufacturer in Article 10. This is also one duty of the person responsible for regulatory compliance with the manufacturer in Article 15. Articles 83 and 84 describe the exact requirements.
What are the Requirements of effective Post Market Surveillance?
PMS is a collection of processes and activities used to monitor the performance of a medical device. These activities aim to quickly uncover design and/or usage problems and accurately characterise real-world device behaviour and clinical outcomes. The need for PMS arises immediately upon commercialisation of the device.
Ensuring adequate medical input during product development will help manufacturers identify safety risks. These activities help define the device’s risk profile and the PMS strategy. Effective PMS requirements should be proportional to the risk associated with the device’s intended use.
Manufacturers should consider whether the product or technology is new to them and/or the market while designing a PMS process. A manufacturer with a long history of developing and marketing similar device types should understand the patient population and foreseeable risk.
Available data regarding state-of-the-art market experience for similar products and technology may be adequate for low-risk devices with a long history of clinical use. These data types often give manufacturers using literature to support clinical evaluation requirements knowledge of the patient population, co-morbidities, and the effect of different patient demographics on the device. High-quality literature (e.g. randomised control trials, meta-analyses) gives manufacturers quantified clinical data on the safety profile of these device types.
Post Market Surveillance requires manufacturers to detail how often key documentation used to certify GSPR compliance will be updated.
You must do this in response to information gained during the PMS. It’s important to note that the PMS combines ‘proactive’ and ‘reactive’ activities which form the basis of the PMS plan.
As part of CE mark certification, you must provide a PMS plan based on clinical data and residual risks.
ISO 13485 applies to all medical devices on the market, so keep that in mind when formulating a PMS plan. In the context of this standard, ‘early warning’ means proactive PMS. We expect post market surveillance to include a PMCF study. If a PMCF study is unnecessary, there should be an adequate rationale for this.
What PMS Reports are Required?
The EU MDR requires manufacturers to prepare post market surveillance reports and periodic safety update reports for medical devices, a requirement previously seen only in pharmaceuticals and biotechnology.
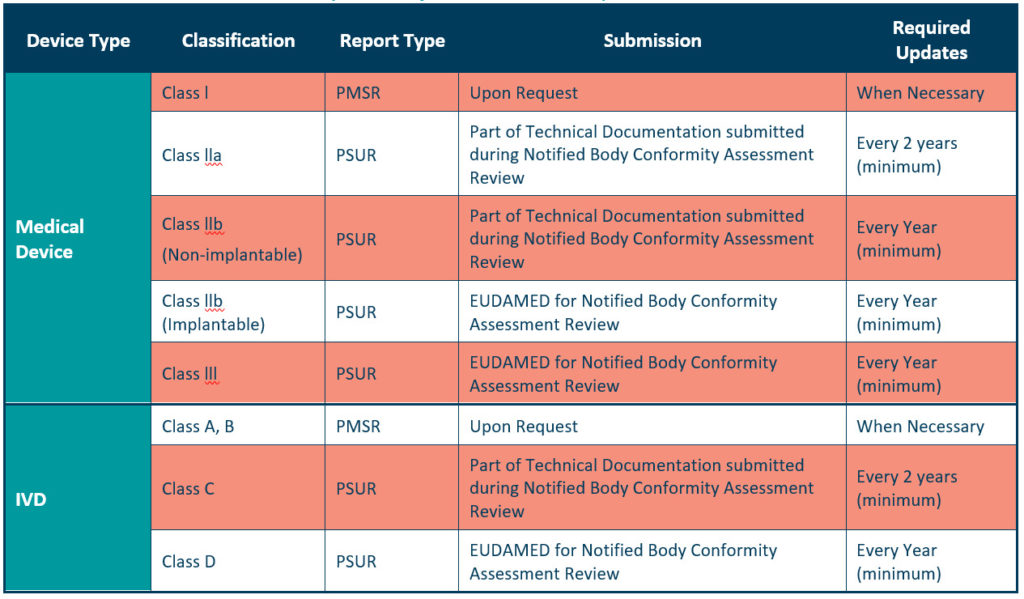
The PMSR applies to low risk, Class I devices. It should summarise the results and conclusions of the Post Market Surveillance data, as well as the rationale behind, and description of, any corrective actions taken for devices on the market. Manufacturers should update the PMSR when necessary, and make it available to the EU Competent Authorities on request, as described in Article 86 of the MDR. We should also update your PMSR when necessary for Class A and B IVDs as listed in the IVDR (see table 2 below, for further details).
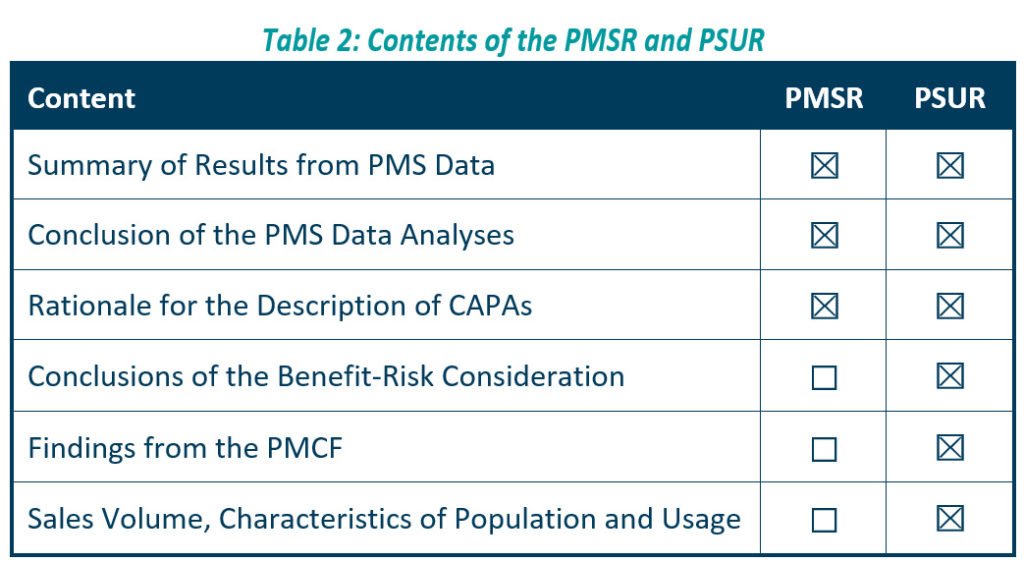
We can consider the PSUR an extension of the PMSR, which contains equivalent information for higher risk devices, i.e., Class IIa, Class IIb, and Class III devices. Like the PMSR, the PSUR requires manufacturers to summarise the results and conclusions of the PMS data. It also requires a summary of post-market information, vigilance reporting, and the current market status of the devices in the EU.
Note that the frequency of the report updates doesn’t correspond to the frequency of data collection and analysis. You should collect data and perform analysis continuously, throughout the lifetime of the device, on a daily, weekly, or monthly basis.
The PSUR is for higher class devices and contains more information than the PMSR.
According to the European Medicines Agency, you need to design the Periodic Safety Update Report to “provide a comprehensive and critical analysis of the risk-benefit balance of the product, taking into account new or emerging safety information in the context of cumulative information on risk and benefits”.
Therefore, the PSUR becomes an important source in the identification of changes to the risk-benefit profile, new safety information, and also an effective indicator of the real-life behaviour of a medical device.
According to article 84 of the MDR, the PSUR should describe the conclusions of the benefit-risk determination, the main findings of the Post-Market Clinical Follow-Up (PMCF), and the volume of sales of the device, as well as characteristics of the population using the device and of the usage itself.
You should submit your PSUR during the Notified Body Conformity Assessment review or through the EUDAMED for Notified Body review.
For Class IIa devices, you must update the PSUR every 2 years at a minimum, while updates are required at least every year for Class IIb and Class III devices.
Vigilance Data Gathering Needs EU MDR
The MDR describes that vigilance procedures clearly differentiate from Post Market Surveillance and is far more extensive than in the MDD. Manufacturers can find vigilance requirements in Chapter VII, Section 2 of the MDR (articles 87-92).
A vigilance plan is a required part of the PMS plan, as defined by Article 83 of the MDR.
Your vigilance plan should include descriptions and methods for:
- Collecting data on serious incidents
- Distinguishing serious incidents from expected side effects
- Analysing serious incidents and the procedures for escalating and notifying competent authorities
- Receiving and analysing complaints
- Compiling data on trends
- Reporting significant data to the appropriate authorities
Article 89 describes how the Vigilance Plan must describe processes for following up on serious incidents.
The follow up on serious incidents should include:
- Investigations of the incident.
- Update on risk assessment.
- Communication with notified bodies and competent authorities.
Manufacturers must include the Vigilance Plan in the technical documentation submitted to the notified body for assessment.
Vigilance Reporting Under EU MDR
Vigilance reporting is the action of reporting to the competent authorities when an action is taken to reduce the risk of death or serious deterioration in health by the manufacturer, such as a recall or a Field Safety Corrective Action (FSCA). The manufacturer or authorised representative (AR) is responsible for distributing the vigilance reports to the competent authorities in countries where the device is being commercialised and where the manufacturer and their AR are located.
Failure to report an FSCA can lead to severe consequences. I think we can all recall one or two vigilance scandals over the last few years, which led to serious medical and legal consequences. The timelines associated with serious incident reporting are largely the same as under the MDD, with the exception of incidents that did not lead to death or serious deterioration.
Table 3: Vigilance Reporting Timelines
Type of Incident | MEDDEV 2.12-1, Rev. 8, Section 5.17 | EU MDR Article 87 and IVDR Article 82 |
Serious Public Health Threat | Report Immediately but not more than 2 Days | Report Immediately but not more than 2 Days |
Death or Unanticipated Serious Deterioration in State of Health | Report Immediately but not more than 10 Days | Report Immediately but not more than 10 Days |
Others (could have led to death or serious deterioration in health) | Report Immediately but not more than 30 Days | Report Immediately but not more than 30 Days |
Table 4: Relevant Guidance and Sections of the Regulations
Section | Article | Regulation |
MDR Article 10 | Article 9 | General Obligations of Manufacturers |
MDR Article 33 | Article 2 | European Database on Medical Devices |
Chapter VII, Section 2 | Article 87 | Reporting of Serious Incidents and Field Safety Corrective Actions |
Chapter VII, Section 2 | Article 88 | Trend Reporting |
Chapter VII, Section 2 | Article 89 | Serious Incidents and Field Safety Corrective Actions |
Chapter VII, Section 2 | Article 90 | Analysis of Vigilance Data |
Chapter VII, Section 2 | Article 91 | Implementing Acts |
Chapter VII, Section 2 | Article 92 | Electronic System on Vigilance and on Post-Market Surveillance |
Chapter VII, Section 3 | Article 93 | Market Surveillance Activities |
CLIN-r+ Recommendations
CLIN-r+ offers Vigilance and effective Post Market Surveillance solutions for any size IVDR or Medical Device manufacturer. This includes:
- Quarterly Report PDF/Doc
- Global Adverse Event Search
- Key AE Reviewed + Highlighted
- Tending Data + Benefit Risk Update
We customise our solutions to our client needs. So, whether you are a new SMA or a team already completing/submitting PSUR and PMCF reports, and looking for supplemental data with which to build those documents.
This package can create an EU MDR compliant PMS system or be a complement outsourced service to remove a significant amount of the labour required of your team to maintain frequent reports/trending data.
To learn more about CLIN-r+ services for medical devices and IVD manufacturers, get in touch!
