You Got CE Marked – What’s Next for your Medical Device?
What’s next for your Medical Device?
The CE mark is a major milestone for most medical devices. It can take a long time to obtain and be a challenging process. After receiving the CE mark, the ongoing QMS audits of your Technical Documents process to demonstrate your products lifecycle compliance with the EU MDR (2017/45) and IVDR (2017/746) begins.
Manufacturers who are new to the MDR might be wondering just how much more work is involved to demonstrate in your compliance in Notified Body inspections. The obligations and documents required are substantially more than they were for MDD (93/42/EEC).
Here we look at the manufacturer responsibilities once the CE mark has been granted. We discuss update requirements, report content, and document update frequency.

General Obligations
Article 10 of the MDR and IVDR describes the general and post-market obligations of medical device manufacturers.
We’ll review these responsibilities in more details below, but there are several more requirements needed. These include:
- Implementing Unique Device Identifiers (UDI-DI) and Production identifiers (UDI-PI).
- Appointing Economic Operators (evidenced in Service Level Agreements) and suitable Person Responsible for Regulatory Compliance (PRRC).
- Updating labelling and device documents such as instructions for use, implant cards (if applicable) and information to be supplied to the patient considering language requirements.
These are also necessary to ensure you maintain a compliant Quality Management System (QMS).
Risk Management
When risk management is done correctly after CE marking, it can significantly reduce Field Safety Corrective Actions (FSCA). This also ensure the safety and wellbeing of the device users and treatment population(s). It also shows that you have considered all the risks related to the use of your device. Which will in turn help to avoid unwanted events once the device hits the market.
What Should Be Included?
In brief, the EU regulation requires the following:
- Any risks associated with the device must be reduced as far as possible.
- Manufacturers must implement a system for risk management and apply it to each device they develop.
- The measures used to control the risks must be state of the art.
- Risks related to human factors must also be addressed.
- The mitigation measures must remain effective throughout the life of the device.
- Risks must not be affected by transporting or storing the device.
- All foreseeable residual risks must be outweighed by the benefits with result from using the device.
These risk management requirements also apply to device like products listed in Annex XVI.
The MDR/IVDR does not specify a harmonised standard for risk management. The most straightforward approach for Manufacturers will be to implement the risk management system described in ISO 14971: Application of risk management to medical devices. This will describe what needs to be done. However, you will still need to determine who undertakes responsibilities and how this is done.
Continuous Risk Management
Risk management must be continuous. It is not enough to just perform risk assessment when developing the device. The MDR states “risk management is a continuous iterative process throughout the entire lifecycle of a device, requiring regular, systematic updating.“
Post-market procedures must collect all relevant data. This should be taken from patients, users, healthcare professionals, the supply chain, publicly available information, literature reviews, safety information, social media, regulatory databases, clinical evidence, etc. This information should be evaluated to determine any previously unknown problems or circumstances. As well as changes to risk acceptability due to increased severity or probability, state-of-the-art changes, or other risk factors. You need to assess and manage any risk changes after evaluation.

Clinical Evaluation
Clinical evaluations for medical and IVD devices consist of verifying safety and performance information, and clinical benefits, when used as instructed in the IFU. This is done by reviewing and summarising clinical evidence. Clinical evaluation demonstrates the device’s risk-benefit profile is equivalent to current treatments or technologies. It must be comparable to the current state-of-the-art.
Clinical data originates from literature analysis, clinical investigations, and post market surveillance on the subject device and similar or equivalent device(s).
When do I need to update my Clinical Evaluation?
The first clinical evaluation report is just the start. After you have your CE mark, you must routinely update your clinical evaluation throughout the lifetime of your device. In general, a CER needs to be updated anywhere from at least annually, up to every 5 years, depending on the risk class and how well-established the device is.
However, if new information that affects the risk-benefit profile of your device is discovered, then you should update the clinical evaluation immediately.
Literature Review updates – Target device and State of the art (SoTA)
Your literature review needs to be updated at the same time as your clinical evaluation report. A systematic review must be undertaken regularly to keep up with new publications on your device but also the state-of-the-art (SoTA), this process must update your expected clinical benefits and risk using new clinical data to provide ongoing evidence to ensure the NB that your device is safe and works as intended.

Post Market Surveillance (PMS)
Post market surveillance ensures that a device is performing as intended and that it remains safe for patients and users. Manufacturers need to set up a proactive and systematic process to continuously collect information.
This needs to continuously gather data on the safety and performance of your device throughout its lifetime after CE marking.
Post market surveillance consists of both proactive and reactive processes:
- ‘Reactive’ PMS – responds after an event, ranging from complaints to major injury or death (vigilance). We call these activities ‘passive’ because they mostly collect data.
- ‘Proactive’ PMS – tries to foresee and prevent problems.
When do I need to update my post market surveillance?
Post market surveillance reports must be updated according to device classifications.
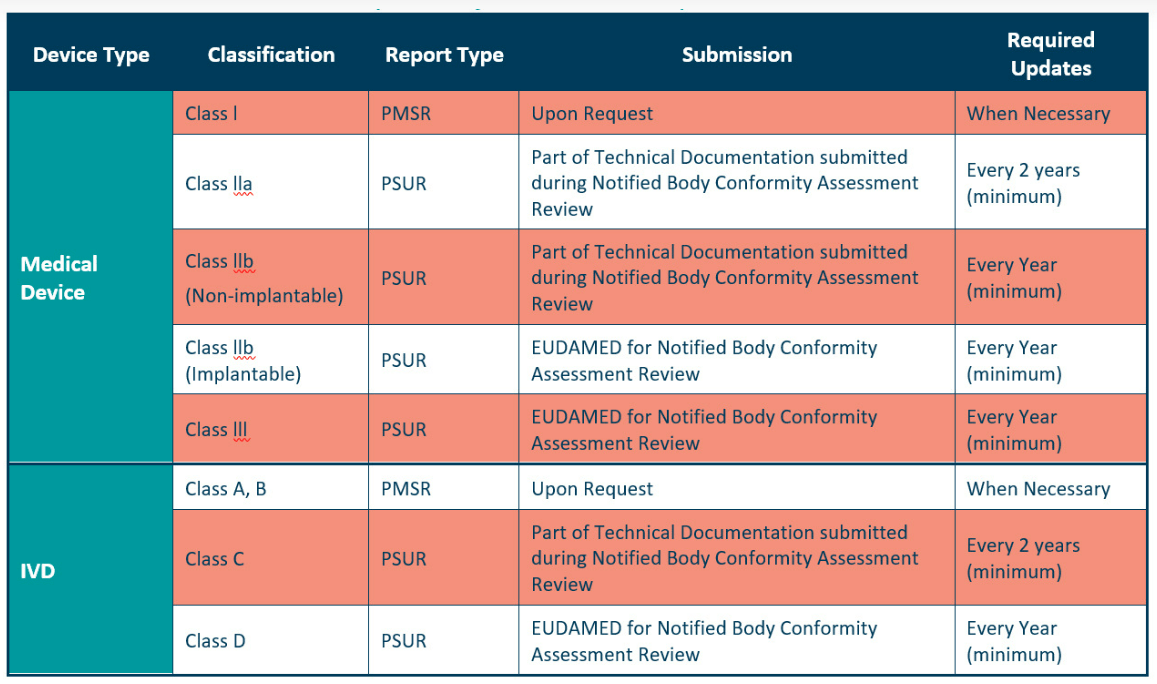
The PSUR is for higher class devices and contains more information than the PMSR.
More in depth information can be found in our Effective Post Market Surveillance paper. However, for convenience the contents are outlined below.

Post Market Clinical Follow Up (PMCF)
Where there are gaps or uncertainty of the clinical benefit or risk of your device which general post market surveillance and clinical evaluation data can’t provide then you require a well-designed post market clinical follow-up study to address this gap. It is described in MDR Annex XIV, Part B, and IVDR Annex XIII, Part B.
Not every device needs PMCF studies but make sure you can justify this against the guidance provided when PMCF activities are not needed. Updates on PMCF studies is highly dependant on your design but it is imperative to plan this in accordance with your auditing activities that you can show data for compliance, even interim data would be accepted.
In the case of high-risk devices annual PMCF report are expected hence carefully designed PMCF plans and suitable study designs that provide periodic updates are essential to account for this. While in lower risk devices that require PMCF reports could be done less often, depending on their residual risks. The MDR Article 83 states that “manufacturers shall plan, establish, document, implement, maintain and update a post-market surveillance system in a manner that is proportionate to the risk class and appropriate for the type of device”. If a Manufacturer doesn’t think their device needs a PMCF, they must provide suitable justification in their post market surveillance plan.
PMCF evaluates your device’s clinical data for safety and performance in order to:
- Identify any previously unknown side-effects or device deficiencies
- Monitor previously identified side effects and contradictions.
- Identify and analyse developing risks.
- Maintain the risk-benefit profile balance.
- Identify potential systematic misuse or off-label device use to ensure the intended purpose is appropriate.
The Medical Device Coordination Group (MDCG) has issued guidance on the PMCF plan and report. The data needed for your PMCF activities varies. General data such as user feedback, information from scientific literature, and other sources of clinical evidence. Specific data such as randomised controlled trials, PMCF studies, or a registry database. Your PMCF plan should justify and document the data you include.
Vigilance
Vigilance is “the identification, reporting and trending of serious incidents and the conduct of safety related corrective actions.” It is the reactive process in post market surveillance.
Manufacturers must report serious incidents to the relevant competent authorities within the following timeframes:
- Serious incident: no later than 15 days after becoming aware of the incident.
- Death or serious deterioration of a person’s state of health: no later than ten days after becoming aware of the incident.
- Serious public health threat: no later than two days after becoming aware of the incident.
MEDDEV 2.12/1 Rev. 8 provides guidance on medical device vigilance systems. There is also additional guidance regarding the Vigilance System as outlined in the above. MEDDEV guidance doesn’t specify the timelines for follow-up. However, submitting a follow up report within 30 days or as soon as additional information becomes available is recommended.
Manufacturers should note that EU member states could have different timeline expectations from the European Commission. So, check the member state’s rules and guidelines where your incident report is submitted.
Technical Documentation Updates
Once the technical documentation is in place and compliant with EU regulations, it must be continuously updated to support your device on the market. It needs to be maintained constantly to accurately reflect the device being manufactured and sold throughout its life.
The EU MDR states that medical device manufacturers must:
- Prepare technical documentation before placing a product on the market.
- Ensure technical documentation is available to the market surveillance authorities if they request it as soon as you place the device on the market.
- Keep records of technical documentation for 10 years from the date you place the product on the market (unless explicitly specified otherwise).
It is worthwhile performing an ‘audit’ like review of your technical documentation at regular intervals to ensure it is kept up to date. We recommend that you create a change log to provide an overview. Manufacturers must note that some changes require notified body approval before they are implemented. Further details on this can be found in Annexes IX and X.
A lot of work is required to keep your technical documentation updated after your CE mark is received. You also need to consider that this must be done for each device on the market.
Clin-r+ recommendations
Risk management documentation and activities need adequate resources. Your device’s safety information should be monitored, evaluated, and resolved quickly.
Clinical evaluation is a crucial part of your technical documentation, so it must be updated routinely.
Post Market Surveillance reports and periodic safety reports must include vigilance information. You need to describe any field safety corrective actions and how they impact your device’s risk-benefit profile.
Technical Documentation should be reviewed regularly and updated whenever a controlled document changes.
You may want to consider outsourcing recurring document updates. Especially given that clinical evaluation and post market surveillance reports need to be updated every 1-5 years. Partnering with an experienced consultancy can be highly beneficial. Consultancies come with specialized resources that can provide extra support where needed and ensure quality work whilst relieving departmental time and effort.
At Clin-r+, we can keep your technical file up to date after you’ve received your CE mark. We offer services in regular updates of literature reviews, clinical evaluation reports, and post market surveillance documentation. Should you have any questions or need professional assistance, CLIN-r+ has a wealth of experience to call upon. Get in touch!