Three Tips for PMCF Planning
Three Potential Problems
The MDR has dramatically elevated the bar for supporting data quantity and quality. It’s critical to keep the pace of preparation activities in Post-Market Clinical Follow-Up (PMCF) planning. Our work with clients has identified many frequent problems, each of which, if not handled, could have serious commercial ramifications. To help manufacturers improve their competitive positioning, this document attempts to highlight those common problems. Plus, it reminds us of the three major best practices steps to compliance.
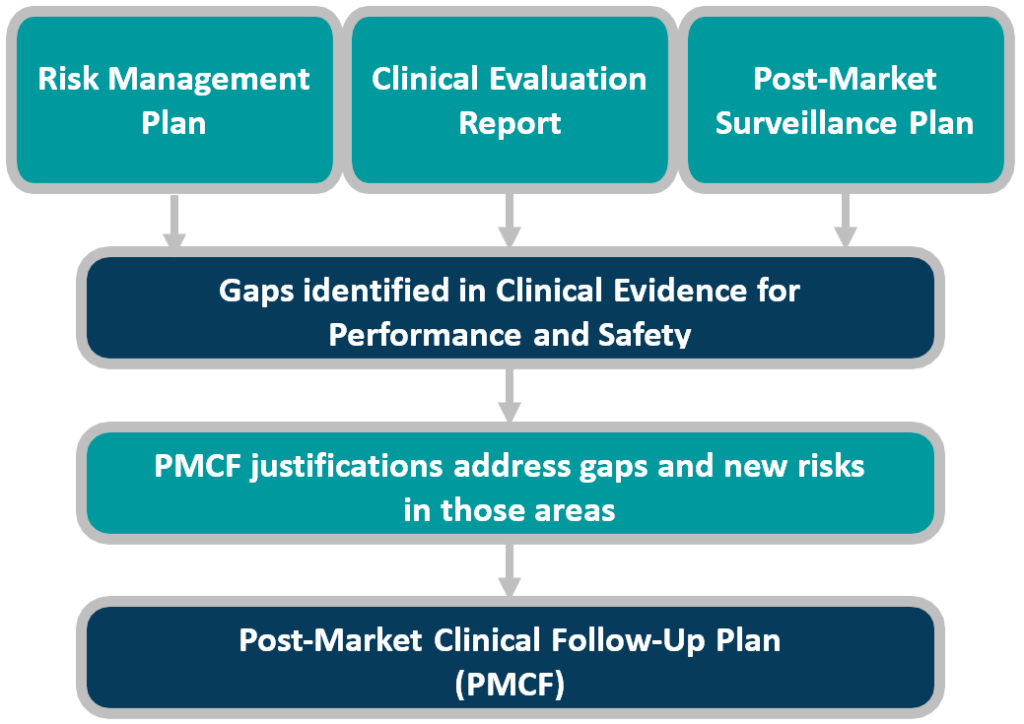
This white paper provides manufacturers with best practice guidance for preparation, steps, and the three key pitfalls which CLIN-r+ frequently encounters. As an overview, Figure 1 reminds us of the process and interdependent processes. Manufacturers must address gaps identified in the CER or emerging risks seen in the PMS process, in the PMCF plan, and in turn, these upstream documents.
PMCF Potential Problem #1:
‘Sufficient Clinical Evidence’
Defining “adequate clinical evidence” is the biggest PMCF compliance hurdle for businesses. When PMCF planning it is critical to recognise that the interpretation of ‘sufficient’ involves a balance of quantity and quality. Do we need 3 patients’ feedback, 30 patients’ feedback, or 300 patients’ input? Is the feedback giving you useful information that you can use to improve your product’s performance?
The device’s risk class, indication, claims, available data to support the device, and any recent changes in clinical practice will all influence the responses to these questions. When evaluating clinical articles as supporting data, ruthless rigour is necessary. We’ve encountered cases where manufacturers were persuaded to provide text that is unquestionably about the product, but does not examine significant results. Analysing outcome data from high-quality studies relevant to the device’s intended use, and comparing the results to similar devices or other therapies, is an important part of determining if the device has enough clinical evidence.
PMCF Best Practice Tip 1: Ensure that Data is Assessed and Identified in a Consistent Manner
Adopt a uniform assessment process for all products – this is the first step in enabling meaningful cross-company communications and comparisons. A uniform data evaluation approach promotes consistency across devices, ensures you do not miss regions when workloads are divided, aids prioritisation in subsequent stages, and presents information consistently for effective comparison.
The strategy should allow clinical data to be arranged in such a way that it is evident what data is available for each device, medical indication, and target group. Before identifying all relevant data, you may need to update the clinical evaluation, so allow time for this.
Contextualise data in a broader risk framework. This could include trends in complaints, severity metrics for complaints, adverse events, recalls/FSCAs/FSNs, sales data, changes in specific marketing regions, political risk, hazard legalities, etc.
PMCF Potential Problem #2:
Collaboration with Other Departments and Workflow Communication
The next big stumbling block we see in our work with manufacturers is a failure to connect early in the process with departments other than clinical, such as sales and marketing or general management. For PMCF performance and continued regulatory compliance, gaining support from other stakeholder business units is critical.
When PMCF planning, cross-functional alignment is important, because this level of examination and clinical evidence applies to both legacy and new products. In fact, legacy devices that have been on the market for a long time are likely to be the most vulnerable to these hazards. Non-specialists in the industry ask, “why do we need to spend money on compliance today for widely established products?” The fact is that the new regulation requires such clinical evidence. Acquiring sufficient data – both in terms of number and quality/relevance – can be expensive. For these operations, it is critical to align on device priority, submission risk, and all accessible sources of data on a product.
We hope it is now clear why it is critical to involve other departments in the process and help them understand why they should care, why they should fund, and the potential business consequences of poor data quality and non-compliance.
PMCF Best Practice Tip 2: Examine All the GSPR Data
- Notified Body reviews will discover clinical data gaps or emerging problems after the manufacturer has collected clinical data for CERs, PMS and Risk Management evaluations for each device.
- Examine data quality and appropriateness. How useful is information gathered, and literature cited? Does the data support the device’s safety, functionality, and intended use? Is this the evidence enough for risk class and the NB?
- How can manufacturers fill the gaps in the most effective/efficient way? Can SOTA, risk level, compliant data, or market experience support them? Should each indicator be more specific? Is there a list of relevant outcomes to support the clinical benefits? Does any bias need to be addressed? What percentage is due to post-market surveillance? How long is product support for? Does the clinical data include all product variations and ranges?
- Assign ownership with a responsibility matrix. Firstly, within product teams (TDs, IFUs, product claims, SOTA). Then across Clinical & Performance, Regulatory Affairs, Product Quality, and Sales & Marketing. Recognise full device-specific requirements exist across the product portfolio. Collaborate with PMS, Marketing, R&D, and General Management. You must allocate resources to each device and prioritise according to certificate expiration date, gap analysis, likely life cycle (Implants), sheer amount of devices requiring data clean-up, and the product’s position within broader therapeutic systems.
- The system must adapt to each product’s specific needs. How would you convey the data to an auditor? How does device data differ in terms of classification, range of indications, innovation, or market history? What are the device class and clinical gaps based options? Are there exceptions to the WET rule? Is a lower data bar acceptable/advisable?
- PMCF planning needs many important commercial decisions, such as whether to keep all current products on European markets. Early starters can use PMCF planning to avoid leaving the market.
PMCF Potential Problem #3:
Documentation is lacking in detail
MDR sets a considerably higher bar for clinical evidence. Even when making minor changes to existing items, such as adding an antimicrobial coating or changing the manufacturing procedure or material.
The third most common PMCF compliance stumbling block is a reluctance to provide adequate detail in the documentation. Despite the efforts of the Medical Device Coordination Group and the Notified Bodies to make the detail abundantly obvious, far too many businesses fail to do so.
It is vital to ensure that the amount of PMCF necessary, as well as the explanation for it, is consistent with the suggested strategy, which is based on good scientific principles, measurable targets, and statistical planning. Document the planned facts and justification, the underlying rationale vs. the hazards, and finally the references (footnotes to EU MDR, guidance, etc). Furthermore, the statistical justifications for the level of detail and data quality supplied are frequently insufficiently robust.
These are the arguments for why the requested volume and type of clinical data is reasonable in light of the device’s dangers. Also, try setting up pre-defined action triggers ahead of time. Finally, if you decide not to do something, you must have the strongest possible justification for your decision.
PMCF Best Practice Tip 3: Develop a PMCF Strategy that is Compliant to EU MDR and ISO 14155
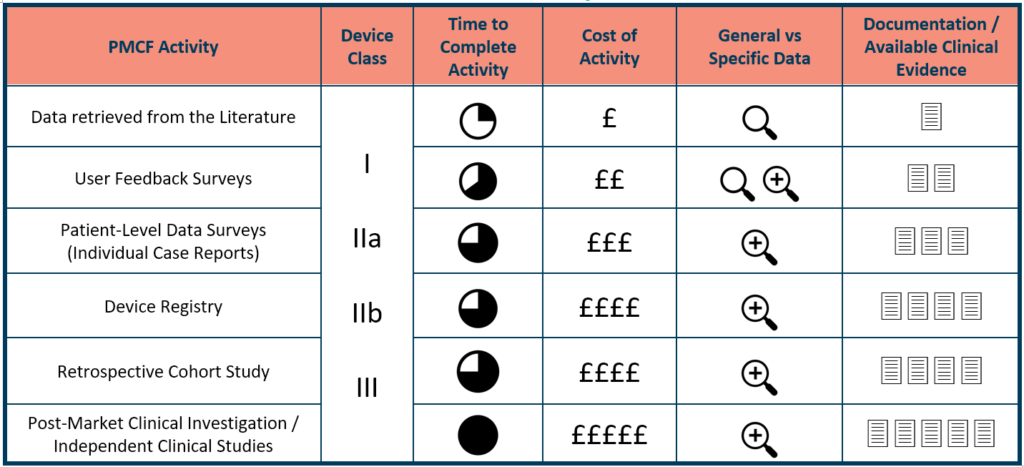
Understand your PCMF strategy alternatives, including the cost and effectiveness of each device type, as well as any gaps. Clinical literature reviews need the least effort and money, followed by patient and customer surveys/questionnaires, clinical database information, and clinical RCTs. All of this contributes to the development of the most robust, meticulously documented reasoning for PMCF data inclusion/exclusion, depth, and relevance.
Recognise the best combination of people, systems, and talents for the most cost-effective solution. What activities should be performed in-house, and which should be outsourced? Even when adequately budgeted, skills and resources are in short supply during times of acute market pressure.
What is the relationship between your PMCF strategy and Notified Bodies? “Consultation or guidance to the manufacturer, the authorised representative, a supplier, or their commercial competitor regarding the design, construction, marketing, or maintenance of the products under examination” is prohibited for NBs.
Have strong justifications for PMCF plans that will withstand examination. Justifications should be based on good scientific concepts and include explicit, measurable goals, as well as extensive documentation. The use of economic rationale (exorbitant costs) to rationalise risk-taking is never a good idea. Each PMCF activity should have its own business considerations. Timeline of the activity, available budget, risk class of the device, quality of data needed to establish sufficient proof, and whether a general or specific PMCF activity will give the data required are all factors we propose include in the evaluation.
CLIN-r+ Recommendations
Partner early with Clinical Affairs consultancy specialised in Clinical Evaluations and PMCF planning to help you tame all your data.
A Clinical Evaluation Matrix (CEM) can organise your data by indication, claim, safety endpoint and performance endpoints. A well formulated CEM saves time by identifying data gaps and state of the art benchmark performance and safety to quickly highlight PMCF data needs.
You will need to team up with various skilled experts.
Including biostatisticians, clinically trained design experts and clinical investigation medical writers, to help clarify the needs of ISO 14155. These professionals should sign off your PMCF plan to demonstrate the expertise sought when PMCF planning. This is an excellent sign of a proficient Clinical Affairs consultancy with expertise available. What design and sample size of patients you would need for PMCF plans? Asking this will quickly highlight vital skill gaps.
Start your PMCF journey by having suitable processes and templates formulated to ensure all required information is formulated and adequate.
With your first PMCF plans, draw and test the process with a few representative and high-priority devices. Then pilot and refine it from the PMCF plan draft to the completion of the PMCF report. This will be used to test templates, forms, and processes to ensure that they perform well across your organisation. It’s also vital to remember that the PMCF process isn’t a one-and-done affair. Manufacturers must consider how the PMCF process interacts with PMS, clinical evaluation, and risk management processes. Alongside how the documents will be updated.
We’ve highlighted potential problems that medical device and IVD manufacturers may face in preparing for MDR compliance (PMCF in particular). Manufacturers can prevent major future financial and market access repercussions by ensuring their business has a solid PMCF plan in place. Preparing for MDR/IVDR in a timely manner (even early, if possible) requires a balance of in-house and outsourced expertise. Medical device manufacturers can build and implement successful PMCF roadmaps by partnering with third-party specialists.
To learn more about CLIN-r+ post market services for medical devices and IVD manufacturers, get in touch!Manufacturers can prevent major future financial and market access repercussions by ensuring they have a solid PMCF plan in place.
