Translating your FDA 510K to an EU MDR submission
What’s the difference between medical device approvals? FDA vs EU MDR explained
Overview
FDA and EU MDR definitions differ. There are significant disparities, despite the fact that under both you can claim equivalence to another device, and many common standards to prove conformity with validations are comparable.
The 21-CFR Quality System Regulations are the foundation for FDA regulations. These are divided by type of device. Like the EU MDR GSPR, performance and safety are prerequisites for all devices, and manufacturers must adhere to their risk profile and preferred market strategy.
The EU MDR is more comprehensive in its instructions. The procedures for device registration, the needs of your Quality Management system, clinical data requirements, the CE certification process, and the obligations of economic operators are all outlined in MDR. It presents post market surveillance (PMS), specifies confidentiality and financial responsibilities, and gives information on data protection. It also explains essential requirements (GSPR) and conditions for notified bodies. In comparison to the FDA, it has different sanctions, financial obligations, and procedures for collaboration.
Device Classification – 510k
FDA categorisation is determined by device risk. The 510(k) standard governs Class I (low risk) and Class II (medium risk) devices. Manufacturers must show that these perform similarly to a device that has already received approval.
Pre-Market Approval is necessary for Class III (high risk) devices, such as implanted devices (PMA). This must prove and validate the device’s entire risk-benefit analysis, performance, and safety.
Device Classification – MDR
The MDR includes the following 4 categories of devices:
- non-invasive devices.
- invasive medical devices.
- active medical devices.
- a special category that includes contraceptive, disinfectant, and radiological diagnostic medical devices.
The MDR classifications are risk based and determine what data and evaluation is required.
- Class I – Non-sterile or no measuring function (low risk)
- Class I – Sterile and a measuring function (low/medium risk)
- Class IIa (medium risk)
- Class IIb (medium/high risk)
- Class III (high risk)
Technical Dossier and Risk Process – 510k
There are various sections required for your Technical Dossier. The structure over the page (Table 1) is organised into sections of the 510(k) that are usually related and typically focus on similar topics that we would recommend as a Technical Dossier structure. In Table 1 we compare the EU MDR Technical Documentation with a 510K Dossier.
Table 1: Comparative Technical Dossier Structure versus EU MDR Technical Document
Sections | EU MDR Technical Document | Sections | 510K Technical Dossier |
|
| Section 1.0 | Cover Sheet Forms |
|
| Section 1.1 | Medical Device User Fee Cover Sheet (Form FDA 3601) |
|
| Section 1.2 | CDRH Premarket Review Submission Cover Sheet |
|
| Section 2.0 | Public Information About Your Device |
|
| Section 2.1 | 510(k) Cover Letter |
Section 1 | Device summary | Section 2.2 | Indications for Use Statement |
Section 1 | Device summary | Section 2.3 | 510(k) Summary |
|
| Section 3.0 | Templated Sections |
|
| Section 3.1 | Truthful and Accuracy Statement |
|
| Section 3.2 | Class III Summary and Certification |
Section 6 | 6.2 Clinical Data validations Declarations of Interest | Section 3.3 | Financial Certification or Disclosure Statement |
Section 4 | 4.1General safety and performance requirements 4.3 Declaration of conformity | Section 3.4 | Declarations of Conformity and Summary Reports |
|
| Section 4.0 | Comparing Your Device vs. Predicate(s) |
|
| Section 4.1 | Executive Summary |
Section 1 | Device summary | Section 4.2 | Device Description |
Section 6.2 | 6.2 Clinical Data validations 6.2.1 Clinical Evaluation Report – Section 5 Equivalence route | Section 4.3 | Substantial Equivalence Discussion |
Section 5 | Risk Management File | Section 5.0 | Ensuring Patient Safety |
Section 2 | Information Provided by the Manufacturer | Section 5.1 | Proposed Labeling |
Section 6.1 | Pre-market Validations | Section 5.2 | Sterilization and Shelf Life |
Section 6.1 | Validations | Section 5.3 | Biocompatibility |
Section 6.1 | Validations | Section 6.0 | Software and Electrically Powered Components |
Section 6.1 | Validations | Section 6.1 | Software |
Section 6.1 | Validations | Section 6.2 | Electromagnetic Compatibility and Electrical Safety |
Section 6.1 | Validations | Section 7.0 | Performance Testing |
Section 6.1 | Validations | Section 7.1 | Performance Testing – Bench |
Section 6.1 | Validations | Section 7.2 | Performance Testing – Animal |
Section 6.2 | 6.2.1 Clinical Investigations | Section 7.3 | Performance Testing – Clinical |
The FDA does not audit Class I devices that do not have a measuring function or are non-sterile. Similar to EU MDR that do audit Class I devices that are either sterile, measuring function or reusable. FDA manufacturers who self-identify may sell their products if the necessary paperwork is provided. (Figure 1).
Class II and higher products need to go through the either the 510(k), de Novo or PMA process, which calls for a clinical testing protocol and Design History File (DHF). But note your FDA submission does not require a Clinical Evaluation like the EU MDR.
The majority of device submissions to the FDA fall within the 510k application category. Clinical testing lasts between 6 and 15 months, depending on the device’s function and nature. The following assessments are carried out for 510(k) submissions following the designation of the device classification:
- Preliminary safety testing
- Risk estimation
- Hazard identification
- Hazard management planning
- Risk mitigation strategy planning
- Risk control analysis
- Risk control effectiveness verification
- Overall residual risk analysis
- Risk/benefit analysis
- Final risk management review
Technical Document and Risk Process – MDR
There are several sections that must be included in your Technical Document (Figure 1). The requirements in Annex II specify the structure and must be used if it applies to your equipment. It also introduces the General Safety and Performance (GSPR) checklist, which requires you to state the regulations that apply to your equipment and any validations or supporting documentation you have to demonstrate compliance.
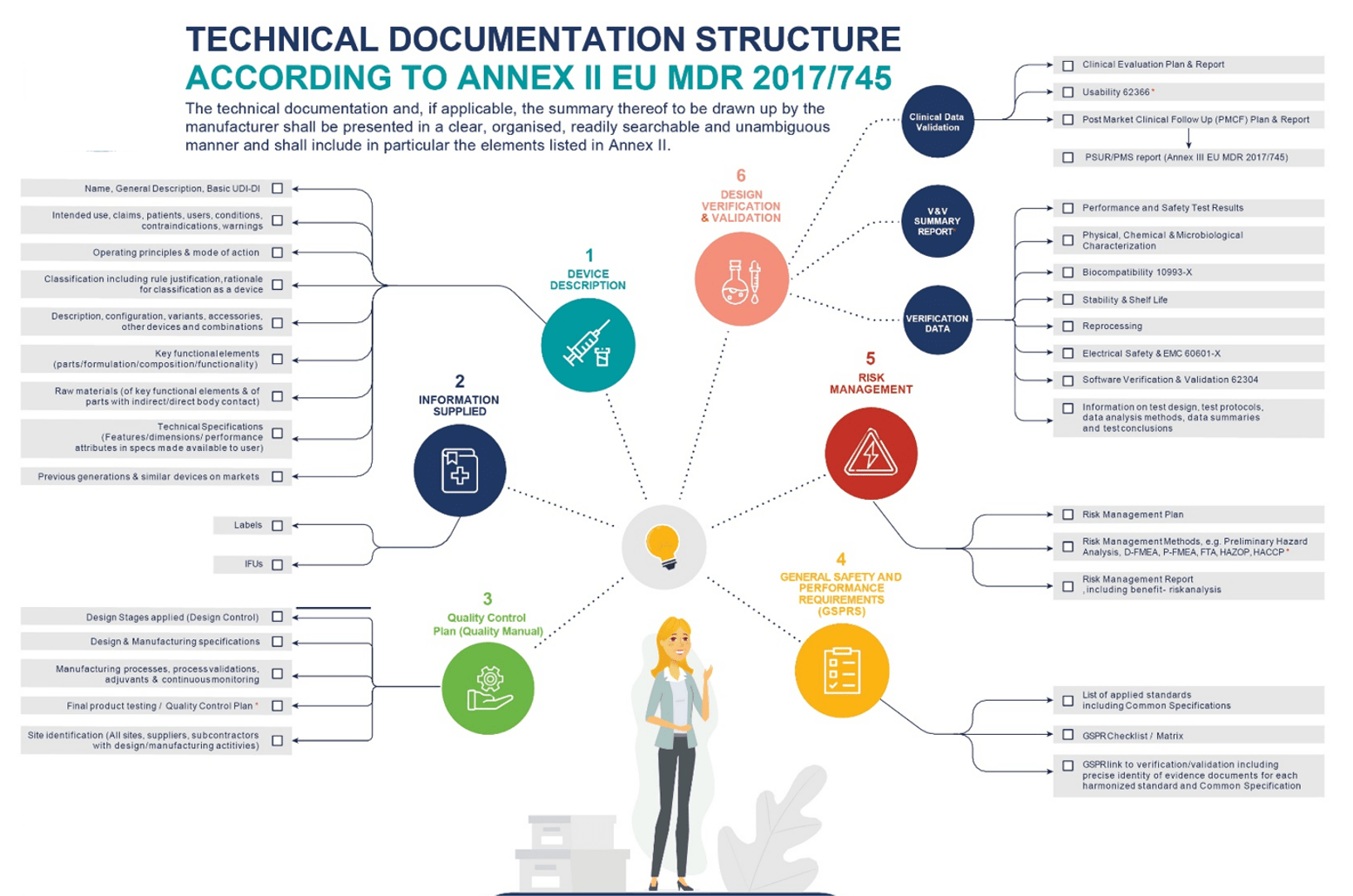
Whilst the majority of Class I and IIa devices require evaluations based on Annex IV and V, they are exempt from conformity assessment to CE marking. Medium-risk devices are subject to conformity assessments in line with Annex XI MDR (Part A).
Comprehensive technical documentation and extensive risk evaluation during conformity assessments with notified bodies (NBs) are necessary for all Class IIb and Class III devices. At least two audits are involved in these assessments. The first reviews the QMS procedures and quality manual for ISO 13485 and associated directives compliance.
The second involves the NB inspecting QMS records and facilities as part of a Technical File review and facility/certification audit. Manufacturers can produce CAPAs and management review/manufacturing documents in between audits to show that the QMS is functioning.
The Clinical Evaluation requirements for EU MDR are a significant departure from the FDA approach. Clinical affairs expertise (medical competence, clinical trial design, biostatistics, clinical research, and medical writing) is required to put together the documentation for the Clinical Evaluations section. The Clinical Development Plan (CDP), Clinical Evaluation Plan (CEP), State of the Art (SoTA), Systematic Review Report, Clinical Evaluation Report (CER), Post Market Clinical Follow-up plan (PMCF), PMSR/PSUR, and SSCP are all included in the Clinical Evaluations section.
Technical Documentation
A Risk Management Report, Design paperwork, and Manufacturing documentation are typically the bare minimum requirements for regulatory processes. A design plan and supporting documentation, including as requirements, architecture, verification, and validation, are required by both the FDA and EU MDR.
Before the certification audit, notified bodies for CE marking demand a design manufacturing report and manufacturing validation.
A Clinical Evaluation Report (CER) is required to be included with the Technical Documentation sent to the Notified Body. The CER is required to identify known hazards and risks by summarising manufacturer data review data, assessing the device’s literature and/or comparable devices, and searching vigilance databases.
Unique Device Identification (UDI)
A set of alphanumeric or numeric characters known as the UDI was developed by an internationally recognised standard. By establishing international standards, it enables clear identification of medical devices sold on the market, improving patient safety.
The FDA specified that each model or version of a device have a unique device identification in both human-readable and AutoID formats in September 2013.
The UDI standards were adopted and refined by the EU MDR in 2017. In order to organise regulated medical devices into EUDAMED, the basic UDI-DI was established, the EU regulatory database for regulated medical devices. The UDI on labels and software must match, which is different from the FDA. Reusable equipment cleaning must also be taken into account.
Connected Medical Devices
When it comes to connected devices, the classification process is the primary distinction between EU MDR and FDA.
The majority of US devices are approved through the 510(k) method; therefore, the classification of a device is established by comparing it to the class of a predicate device. When there is no predicate, FDA approval is more comprehensive, either via the PMA or De Novo route.
All connected devices are categorised by the EU using rules. MDSW (medical device software – known as SaMD in the EU) includes 22 waterfalling rules to follow for device classification. In addition, the US and EU are both members of the IMDRF, and other rules for connected devices are mostly unified.
Can you use the same documentation for MDR and 510k?
The answer to this is yes and no. The bulk of the requirements for FDA and EU MDR regulations are the same. You will be required to provide similar risk criteria, have a Quality Management System (ISO 13485:2016) in place, do PMS, perform validations, and provide safety and performance data.
The requirements and report formatting have a few small differences. You can easily comply with both sets of regulations if you have the current ISO 13485:2016 and a robust, current QMS. The best electronic QMS systems enable you to quickly dump important data into the right places for compliance and are intended for regulatory compliance.
Classify each device first. This establishes your pathway and requirements. Early QMS implementation can help shorten the time it takes for your submissions to reach the market.
The FDA’s MDSAP (Medical Device Single Audit Program) harmonises international inspection standards. but at the moment, the EU is a spectator.
Medical Device Compliance for FDA & EU MDR
FDA and EU MDR medical device compliance is a challenging and ongoing process that necessitates regular inspection and upkeep. Similar tactics are being used by guidelines to maintain device standardisation. For instance, European regulations are advancing toward a tougher evaluation and audit of QMS and product information, similar to the 510(k) methods used by the FDA. Both regulations examine clinical data, product information, performance testing, labelling, benefit-risk analyses, residual risks, and post-market surveillance. To meet requirements for device submission, manufacturers must keep up with evolving regulations.
CLIN-r+ Recommendations
Medical device manufacturers that wish to enter both the US and EU markets, need to fully understand the similarities and differences between the FDA and EU MDR.
Starting a EU MDR gap analysis of your 510K submission early on will speed up your progress in both markets. An experienced regulatory consultant can be invaluable not only in highlighting the gaps but refining your Clinical Development Strategy and providing your team with an infrastructure to compile all the documents to meet the auditors assessment checklist.
Should you have any questions or need professional assistance, CLIN-r+ has a wealth of experience in 510K to EU MDR translations to call upon. Get in touch!
