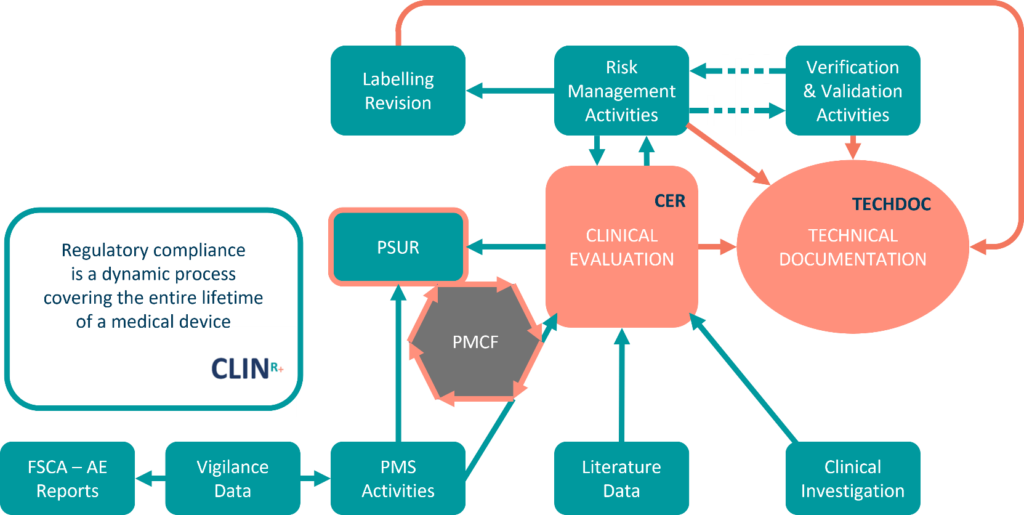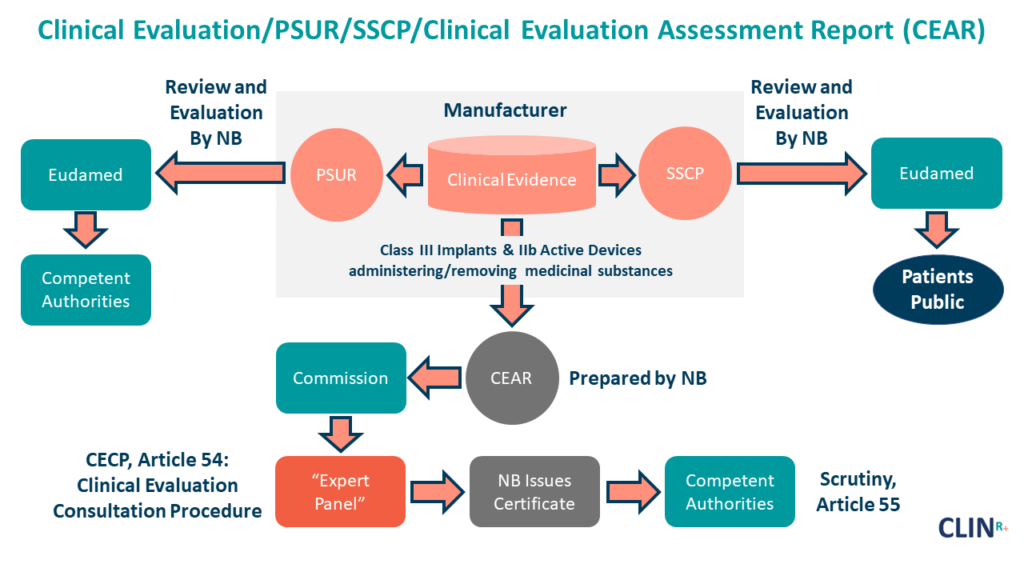
The Clinical Evaluation Report (CER), an evaluation and conclusion of the clinical data collected for your device, is a crucial piece of the Technical Document for conformity assessment and subsequent CE marking in Europe.
The CER is designed to demonstrate the safety and efficiency of the medical device, whilst delivering on clinical performance. Moreover, a Clinical Evaluation Report documents both the assessment and analysis of clinical data collected, with the purpose of verifying the clinical safety and effectiveness of the device.
The report consists of a clinical investigation of the device itself and/or existing clinical studies for comparable devices.
Article 61 of the MDR:
 “A clinical evaluation shall follow a defined and methodologically sound procedure based on the following:
“A clinical evaluation shall follow a defined and methodologically sound procedure based on the following:
- Critical evaluation of the relevant scientific literature currently available relating to the safety, performance, design characteristics, and intended purpose of the device.
- A critical evaluation of the results of all available clinical investigations.
- A consideration of currently available alternative treatments options for that purpose, if any.”
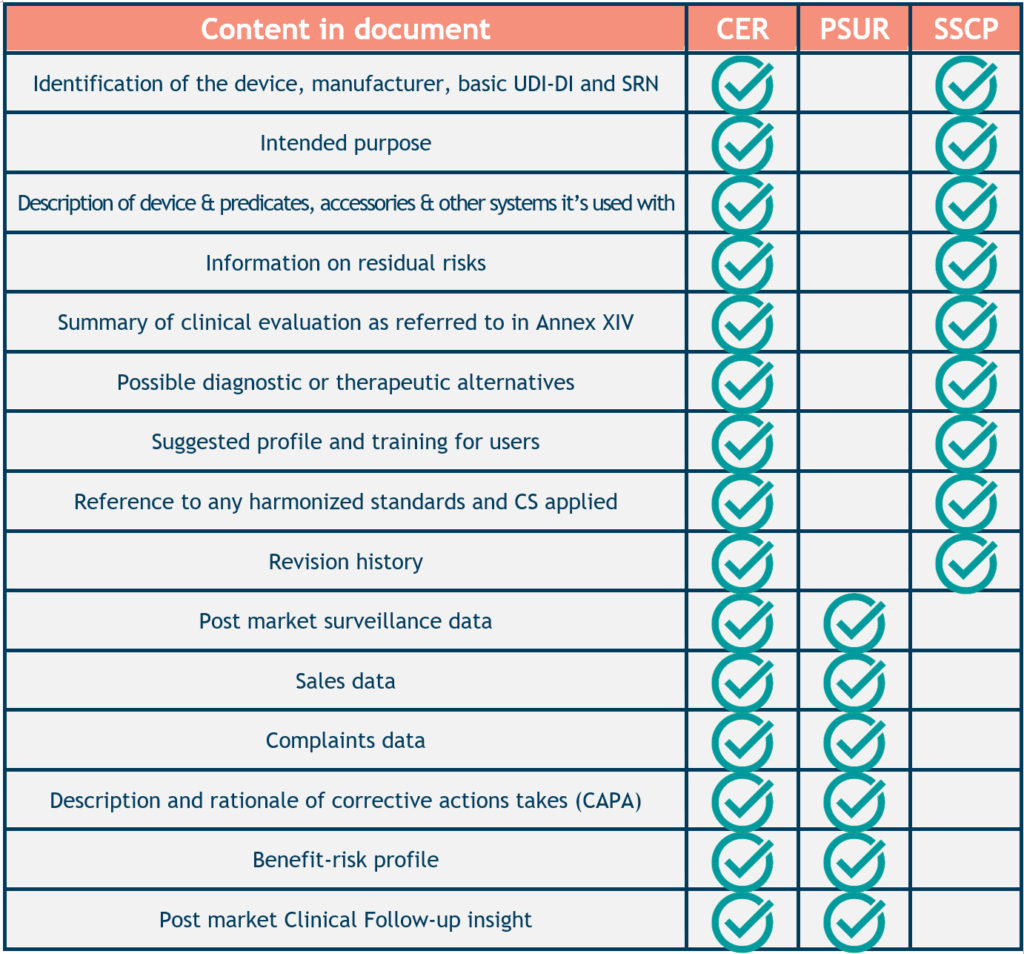
Considering the vast array of medical devices, composing a definitive list of what should be included in the CER is difficult. Higher risk devices pose a greater potential threat to a patient’s health if they malfunction. Therefore, they require a more detailed CER than lower-risk devices.
Although no two CERs are the same, they must all contain certain elements:

- Device information, such as intended use, medical device design, manufacturer name, regulatory history, and any other relevant data.
- Technical device description, such as intended use and target population, classification, therapeutic or diagnostic claims, clinical background, etc.
- Any identified clinically equivalent device and a justification of its equivalence.
- Existing clinical investigation and data.
- Summary of clinical data and review, including documented systematic literature searches and reviews.
- Data appraisal methodology and parameters.
- Conclusions about safety, performance, and conformity, as well as benefit-risk determination.
PSURs are well-known in the pharmaceutical world, but they’re a new requirement in the medical device industry.
Due to a growth in unmonitored medical devices, the medical device sector anticipated the EU MDR will focus more on post-market surveillance data systems. The MDR did not disappoint, emphasising device safety over its lifetime. Post market surveillance is a critical part of a medical device manufacturer’s obligations and regulation.
Article 86 of the MDR mandates the Periodic Safety Update Report.
“Throughout the lifetime of the device concerned, the PSUR shall set out:
- The conclusions of the benefit-risk determination.
- The main findings of the PMCF.
- The volume of sales of the device, and an estimate of the size and other characteristics of the population using the device, and, where practicable, the usage frequency of the device.”
It is imperative that the report be updated continuously throughout the lifetime of the device, from the moment it is put on the market. The Periodic Safety Update Report is an integral part of the technical documentation submitted to the notified body during the conformity assessment.
Not a repetition of the PMSR, the PSUR is a distinct document underlining the safety of the medical device in question. Ordinarily, it would include the following elements:
- Conclusions on benefit-risk analysis.
- Findings from the post-market clinical follow-up.
- Sales volume and usage frequency of the device.
- Vigilance data.
- Descriptions and rationales for any preventive and/or corrective actions made.
- Results and conclusions on the post-market surveillance data.